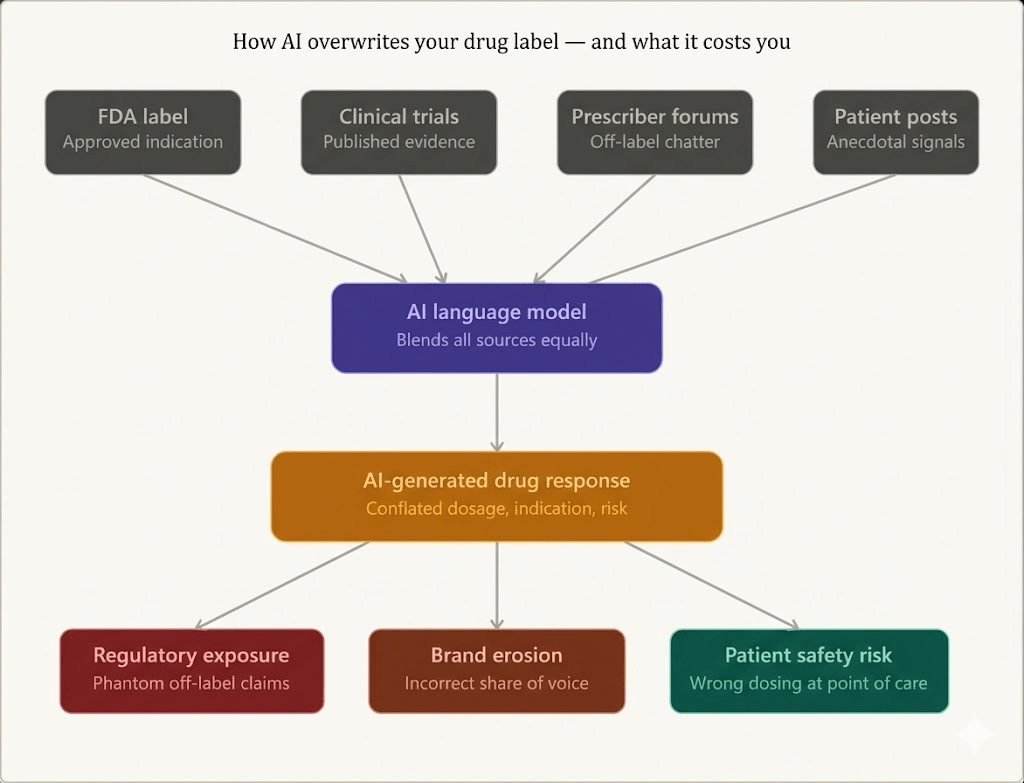
Pharmaceutical brands are losing control of their product narrative — not in the press, not at the pharmacy counter, but inside the AI chatbots that millions of patients and clinicians now consult before anyone speaks to a human being.
The New Front Door to Drug Information
Patients used to call their doctor. Then they Googled. Now they ask ChatGPT, Claude, Gemini, or Perplexity — and they expect an answer in seconds, synthesized from across the internet, delivered with the quiet confidence of a knowledgeable colleague.
That shift has consequences the pharmaceutical industry has been slow to reckon with. An FDA-approved label represents years of clinical development, regulatory negotiation, and pharmacovigilance. It defines an approved indication, a dosing range, a contraindication list, a black box warning. It is, in the most literal regulatory sense, the truth about your drug.
AI doesn’t read it that way.
Large language models (LLMs) are trained on the web — which means they ingest your FDA label alongside Reddit threads, off-label case reports, two-year-old prescriber blog posts, patient testimonials, and clinical forum discussions where physicians speculate about emerging uses. The model doesn’t discriminate by source authority. It blends. It summarizes. And when a patient types ‘what is [drug name] used for?’ into any major AI assistant, the answer they receive may bear only a passing resemblance to what your regulatory affairs team spent years defending.
This isn’t a hypothetical risk. It is happening right now, at scale, to virtually every drug with a meaningful digital footprint.
When AI Becomes the De Facto Prescribing Reference
There is a useful analogy in what happened to Wikipedia in the mid-2000s. Pharmaceutical companies watched, sometimes in horror, as encyclopedia articles about their drugs became primary sources for patients and, quietly, for some clinicians. The difference with AI is velocity and personalization. Wikipedia showed everyone the same article. AI generates a custom answer for each query, shaped by the precise phrasing of the question, the model version, the training cutoff, and whatever retrieval-augmented sources the platform chooses to surface.
The practical implication: two patients asking nearly identical questions about the same drug can receive meaningfully different answers. One might get the correct dosing range. Another might get a response that reflects a clinical trial population the drug was never ultimately approved for. A third might receive an answer that conflates their branded drug with a biosimilar competitor, or worse, with a drug in a different therapeutic class that shares part of its name.
Consider what this means for rare disease drugs. A biologic approved for a narrow patient population — say, a specific genetic subtype of a lysosomal storage disorder — sits inside a training corpus alongside research papers exploring its potential use in adjacent conditions, patient advocacy materials using approximate language, and conference abstracts from investigators testing off-label protocols. The AI model sees all of it. Its output reflects the aggregate, not the label.
Pharma companies tracking brand perception have traditionally monitored media coverage, social listening platforms, and physician surveys. None of those channels captures what happens inside an AI conversation. And because AI responses are ephemeral — they aren’t indexed, they don’t have URLs, they leave no persistent record — the monitoring gap is profound.
The Anatomy of an AI Drug Misinformation Event
How labels get distorted
AI drug misinformation rarely looks like fabrication. The model doesn’t invent a drug from scratch. It takes real information and reconstitutes it in ways that are often plausible-sounding but clinically wrong.
The most common patterns fall into four categories.
The first is indication drift. A drug approved for moderate-to-severe plaque psoriasis may be described by an AI as useful for ‘inflammatory skin conditions broadly’ — a characterization that seems consistent with the mechanism of action but blurs the actual approval boundary. For a company defending its label against off-label promotion scrutiny, that framing matters enormously.
The second is dosing conflation. Where a drug has different dosing protocols for different patient populations — weight-based pediatric dosing versus adult fixed dosing, loading doses versus maintenance doses — an AI that generates a single simplified answer has already introduced clinical risk. The model may average across what it has seen, producing a number that is neither the approved adult dose nor the approved pediatric dose, but something in between.
The third is competitive contamination. In a crowded therapeutic area, the training data includes extensive discussion of competing drugs. An AI asked about Drug A may incorporate attributes, trial outcomes, or adverse event profiles from Drug B or Drug C, especially if those drugs share a mechanism of action or are commonly mentioned together in the clinical literature. The brand distinctiveness that your marketing team has spent years building collapses inside the model’s synthesis.
The fourth is temporal lag. AI models have training cutoffs. A drug that received a label update — say, a new approved indication, a revised contraindication, or a modified REMS requirement — may not have that update reflected in the model’s responses for months or years. For drugs in fast-moving therapeutic areas like oncology or rare disease, where label updates can be clinically decisive, this lag is not a minor inconvenience.
<blockquote> ‘By 2026, 30% of people in advanced economies will consult an AI model before or instead of speaking to a human health professional for common health questions.’ — Gartner, Health Technology Predictions, 2024. </blockquote>
Regulatory Risk Is Not Theoretical
The question pharmaceutical legal and regulatory teams are beginning to ask — and that the FDA has not yet definitively answered — is this: when an AI system misstates a drug’s indication, does the manufacturer bear any responsibility?
The current regulatory framework offers no clean answer, and that ambiguity is itself a risk.
FDA’s guidance on digital health, social media, and online drug promotion has always struggled to keep pace with technology. The agency’s existing frameworks for Internet/Social Media Platforms were designed with human-generated content in mind — a manufacturer’s promotional material, an employee posting to LinkedIn, a company-sponsored website. They were not designed for the scenario where a third-party AI system, trained on publicly available data, makes statements about a drug that the manufacturer has no control over and may not even be aware of.
But here is where the risk calculus gets complicated. If an AI consistently misrepresents an approved drug’s indication in a way that appears to mirror unmet commercial needs — if it consistently describes a drug as useful for conditions adjacent to its approval that the company has been quietly pursuing — regulators may scrutinize the totality of the information environment. They may ask whether the manufacturer’s own digital communications, scientific publications, or congress presentations seeded that perception. The AI is not liable. The manufacturer may still be.
Beyond FDA, there is the European Medicines Agency to consider. EU regulators have historically taken a stricter view of off-label communication. As AI assistants become more prevalent in European clinical settings, the question of how AI-driven drug information fits within the EU’s pharmaceutical promotion rules is a genuine open question for regulatory affairs teams operating across jurisdictions.
This is not merely speculative. In 2023 and 2024, several pharmaceutical legal teams began including AI-generated drug information audits in their quarterly compliance reviews. They were asking what AI says about their drugs, not as a curiosity, but as a formal risk management exercise.
Brand Share of Voice Has a New Dimension
Pharmaceutical brand teams have spent decades measuring share of voice — the proportion of category conversation occupied by their brand versus competitors. The measurement methodologies were built for auditable channels: media monitoring, prescription data, physician surveys, paid media impression tracking.
AI-mediated conversations are none of those things.
When a patient using a major AI assistant asks ‘what’s the best medication for treatment-resistant depression?’ and the AI responds with a ranked or synthesized answer, that response shapes prescriber behavior and patient expectations in ways that no existing share of voice methodology captures. The brand that dominates clinical literature may be mentioned third. The brand with the most aggressive congress presence may not appear at all. The model weights factors that brand teams have no visibility into and cannot directly influence.
Voice of the customer — the real-time signal of what patients and prescribers want, expect, and experience — is increasingly being expressed not through focus groups or patient surveys but through AI-mediated queries. When a patient asks their AI assistant why they experienced a particular side effect, or whether a drug interaction they read about online applies to their situation, that interaction carries diagnostic value for the brand team. It reveals the informational gaps, the anxiety points, and the unmet needs that shape the patient’s relationship with the drug. Currently, most of that signal is invisible.
The companies that figure out how to monitor and respond to AI-mediated brand signals will have an intelligence advantage over those still relying exclusively on traditional social listening and prescriber surveys.
What ‘AI Share of Voice’ Actually Means
It helps to be precise about what pharmaceutical brand teams are actually measuring when they talk about AI mentions.
A drug can appear in an AI response in several distinct modes. In the first mode, it is the primary subject — the patient asked about the drug by name, and the AI is describing it. In the second mode, it appears as part of a competitive landscape response — the AI is describing a therapeutic category and mentioning multiple drugs. In the third mode, it appears as a comparison point — the AI is discussing another drug and references this one for contrast. In the fourth mode, it is absent — the AI discusses the therapeutic area without mentioning the drug at all, which is its own form of competitive signal.
Each mode requires a different response. A primary-subject response that contains dosing errors requires a different remediation strategy than a competitive landscape response in which the drug is described with an outdated efficacy profile. Absence from a category discussion may indicate that the drug’s digital presence, its clinical publication record, or its patient community footprint is insufficient to generate consistent retrieval by AI systems.
Tracking these categories systematically requires querying multiple AI platforms with structured prompt protocols, recording and analyzing the responses, comparing them against approved label language, and repeating the exercise on a cadence that reflects the rate of model update. This is labor-intensive if done manually. It is the core of what platforms like DrugChatter are built to automate — systematic monitoring of how AI systems represent pharmaceutical brands across a range of query types, with comparison against approved label language and competitor mentions.
The Off-Label Problem Is Especially Acute
Pharmaceutical companies are prohibited from promoting their drugs for off-label uses. The legal architecture around this prohibition is extensive and the enforcement history is significant — billions of dollars in DOJ settlements, FDCA criminal charges, and consent decrees over the past two decades have made off-label promotion one of the most closely monitored risk areas in the industry.
AI does not know this.
An LLM trained on the biomedical literature will have ingested extensive academic discussion of off-label uses — case reports, retrospective studies, meta-analyses, investigator-initiated trial reports, prescriber forum discussions. When queried about a drug, the model may incorporate these off-label discussions into its response alongside or even in place of the approved indication. This creates a situation where patients and clinicians are receiving AI-generated information about off-label use of a drug, framed in the neutral authority of an AI assistant, that would be illegal for the manufacturer to have published directly.
The manufacturer’s compliance team did not create this situation. But they are sitting closest to the regulatory blast radius if it becomes a problem.
The obvious partial solution is stronger signal — ensuring the AI models are trained on, or retrievably indexed to, authoritative prescribing information. But pharmaceutical companies do not control AI training pipelines. They can submit structured product labeling to FDA’s DailyMed and ensure it is publicly accessible. They can publish comprehensive prescribing information on their brand websites using structured metadata. They can actively monitor what AI says about their drugs. What they cannot do is inject their preferred framing directly into a model’s weights.
This limitation makes monitoring more, not less, important. If you cannot control what the model says, you need to know what it says.
Clinical Trial Data and the Misrepresentation Risk
Clinical trial results are among the most complex and easily distorted information in the pharmaceutical space. Effect sizes, confidence intervals, patient population characteristics, subgroup analyses, comparator arms — these details matter enormously to clinical interpretation, and they are precisely the kind of nuance that AI synthesis tends to flatten.
A Phase 3 trial that showed a 20% relative risk reduction in a highly selected patient population may be described by an AI as showing that a drug ‘reduces risk by approximately 20%’ — without the qualification that this was in patients who were biomarker-positive, treatment-naive, and without significant comorbidities. For a drug whose market positioning depends on real-world clinical differentiation in a complex patient population, that simplification is commercially damaging. For a patient with the comorbidities that were an exclusion criterion, it may be unsafe.
The post-marketing surveillance implications are equally significant. Drug manufacturers are required under FDA’s pharmacovigilance obligations to monitor reports of adverse events and safety signals. AI-generated responses that mischaracterize a drug’s safety profile — understating known risks, omitting black box warnings, or conflating the drug’s adverse event data with that of a different agent — could, in principle, dampen real-world adverse event reporting by patients who were told by an AI that a symptom they experienced is not associated with the drug.
This is speculative at the margins, but the directional concern is sound: if AI becomes a significant source of drug safety information for patients, the accuracy of that information has pharmacovigilance implications.
The Biosimilar Dimension
The biosimilar landscape introduces a specific category of AI misrepresentation risk that brand teams in biologics are only beginning to map.
Reference biologics and their biosimilars share generic names, share mechanisms of action, and are frequently discussed together in the clinical literature. For a reference biologic manufacturer defending its market position against multiple biosimilar entrants, AI conflation — where the model blends the branded drug’s clinical profile with biosimilar data or attributes — is a direct threat to the brand differentiation argument.
Consider a reference biologic for which the long-term safety data is more extensive than for biosimilar entrants, a common situation given the timeline differences in approval. An AI that summarizes the drug class without distinguishing between the reference product’s safety database and the more limited post-market data for biosimilars is giving patients and prescribers an inaccurate picture of comparative risk. That inaccuracy tends to favor the biosimilar position — it implies an equivalence in safety evidence that does not exist.
For manufacturers of reference biologics, monitoring AI responses for this specific pattern — conflation of reference product data with biosimilar data — is as commercially important as any traditional competitive intelligence activity.
The Signal Value in AI Queries
There is a dimension to this problem that is not purely defensive. AI query patterns carry useful commercial intelligence.
When patients query AI about a drug, the questions they ask reveal their concerns, their misconceptions, and their unmet informational needs. A patient who asks ‘why does [drug name] cause so much hair loss?’ may be experiencing a side effect that the drug’s REMS program or patient support materials address inadequately. A patient who asks ‘can I take [drug name] if I have kidney disease?’ may be in a population for whom the label has specific guidance that is not reaching them through conventional channels.
Aggregated across thousands of queries, these patterns constitute a form of real-world patient insights that pharmaceutical companies would historically have paid substantial sums to collect through market research. They tell you what patients believe about your drug, what they fear, what they don’t understand, and what information gaps exist between your approved labeling and the patient’s lived experience.
The catch is that accessing this signal requires systematic monitoring infrastructure. Individual AI conversations are not aggregated or shared. But a platform designed to systematically probe AI systems with structured query batteries — covering indication, dosing, contraindications, side effects, competitive comparisons, and patient population applicability — can generate a representative picture of how those AI systems represent a drug, and by extension, what the patient who queries those systems is likely to receive.
DrugChatter is one of the emerging platforms purpose-built for this use case, providing pharmaceutical brand and regulatory teams with structured intelligence on how AI systems discuss their drugs, how that representation compares to approved labeling, and how it shifts over time as models update and training data evolves.
Why Traditional Monitoring Falls Short
Pharmaceutical companies have invested heavily in monitoring tools over the past decade. Social listening platforms, adverse event mining tools, media monitoring services, patient community analytics — the monitoring stack is substantial and, in many companies, expensive.
None of it was built for the AI generation of drug information problem.
Social listening platforms monitor indexed public content. AI conversations are not indexed. Media monitoring captures what journalists write about drugs. AI responses are not journalism. Adverse event mining looks for safety signals in specific databases. AI-generated drug misinformation is not captured in FAERS.
The gap is structural, not just a feature gap in existing tools. Monitoring AI-generated drug information requires a fundamentally different methodology: active, systematic querying of AI platforms using structured natural language prompts, across multiple model versions and platforms, on a regular cadence, with outputs compared against a ground truth — the approved label. It is more like audit methodology than monitoring methodology.
The companies that have started doing this have quickly discovered that the gap between what AI says about their drugs and what the label says is rarely zero. In some therapeutic areas, particularly complex mechanisms of action drugs, rare disease agents, and drugs with narrow therapeutic windows, the discrepancies are clinically significant.
The companies that have not started doing this are making a bet — probably unconscious — that the AI information environment about their drugs is accurate and stable. The evidence suggests this bet is frequently wrong.
What Regulatory Affairs Teams Should Do Right Now
Establish a baseline. Query ChatGPT, Claude, Gemini, and Perplexity with structured prompts covering your drug’s approved indication, dosing, contraindications, known adverse events, black box warnings, and patient population. Document the responses. Compare them against approved prescribing information. This is your AI label accuracy baseline.
Track the delta over time. AI models update. Retrieval-augmented systems update their source material. A baseline audit conducted today may be materially different from the output six months from now, especially if your drug has received a label update, if significant new clinical data has been published, or if a competitor has launched. Cadenced re-audit is the only way to track the evolution of the AI information environment.
Map your off-label exposure. For drugs with known off-label use patterns, specifically query AI about those uses. Document how AI systems describe the off-label use — whether they acknowledge it as unapproved, whether they overstate the evidence base, whether they describe it in terms that mirror your drug’s mechanism of action in potentially misleading ways. This is input for a regulatory risk register.
Brief your medical affairs team. In an environment where HCPs are increasingly using AI tools for quick clinical reference, the question of what AI tells physicians about your drug is as important as what your medical affairs team tells them directly. Medical affairs teams should be aware of the AI information environment and should factor it into their educational programming.
Engage on structured labeling accessibility. Ensure your prescribing information is accessible in formats and locations that AI systems can retrieve. DailyMed submission is necessary but not sufficient. Structured, machine-readable prescribing information on your brand website, with appropriate schema markup, increases the probability that AI retrieval systems will surface authoritative information.
The International Dimension
The AI monitoring challenge is not confined to the US regulatory environment.
In the EU, the European Medicines Agency publishes European Public Assessment Reports (EPARs) and Summary of Product Characteristics (SmPCs) for approved drugs. These are the EU equivalents of the US label. AI systems trained on international biomedical literature will have ingested a mixture of US, EU, and other national labeling documents — which frequently differ from each other in approved indications, dosing recommendations, and contraindications.
For a drug approved for a slightly different indication in the US than in Europe — a common situation in oncology, rare disease, and immunology — an AI system may present a confused hybrid of the two labeling regimes to a patient who is asking in a European context. This is not merely a brand management issue. It is a regulatory compliance issue in jurisdictions where the drug is not approved for the use the AI is describing.
Japan, Canada, Australia, and other major markets each have their own labeling standards and their own regulatory positions on digital health information. A pharmaceutical brand team operating in multiple markets needs an AI monitoring capability that can assess AI responses against each jurisdiction’s approved labeling, not just the US FDA label.
The Precedent From Search Engine Optimization
Pharmaceutical companies have navigated a version of this problem before, and the experience is instructive.
When Google became the primary channel through which patients researched drugs, pharmaceutical companies initially treated it as an uncontrollable external risk. Over time, they developed a set of capabilities: rigorous management of their branded digital properties to ensure they ranked highly for branded search terms, investment in structured data markup to ensure search engines retrieved authoritative product information, monitoring of organic search results for drug misinformation, and programs to identify and flag misleading health claims in high-ranking content.
None of that was effortless or inexpensive. It required collaboration between medical affairs, legal, regulatory, and digital marketing. It required new technical capabilities and new monitoring workflows. But it became a standard component of pharmaceutical brand management.
AI represents a qualitatively similar disruption — a new information channel that patients and clinicians trust, that operates largely outside the direct control of the manufacturer, and that requires new capabilities to monitor and manage. The parallel is not perfect. AI is not a search engine, and the mechanisms of AI drug information propagation are different from SEO. But the organizational response — building monitoring capabilities, establishing baseline data, briefing regulatory and compliance teams, and investing in authoritative digital content — follows a similar logic.
The companies that built serious SEO and digital monitoring capabilities in 2012 were better positioned in 2015. The companies that build serious AI monitoring capabilities in 2025 will be better positioned in 2028.
The Patient Safety Case Is the Strongest Case
All of the regulatory and commercial arguments above are real, but the patient safety argument is where pharmaceutical companies should focus their internal advocacy for AI monitoring resources.
AI-generated drug misinformation is not primarily a brand problem. It is a safety problem that creates brand and regulatory consequences. If a patient takes an incorrect dose of an anticoagulant because an AI assistant gave them dosing information that conflated the adult dose with a different formulation, the downstream harm is physical, not reputational. If a patient with a contraindicated comorbidity is told by an AI that a drug is safe for them based on outdated or incomplete information, the risk is clinical.
These are not purely theoretical scenarios. Emergency physicians, pharmacists, and clinical pharmacologists are already reporting cases where patients cite AI-sourced drug information that is incorrect. As AI assistants become more integrated into the patient journey — embedded in patient portals, insurance apps, wearable companion applications, and healthcare-adjacent consumer technology — the frequency of these events will increase.
Pharmaceutical companies have pharmacovigilance obligations that are among the most extensive in any regulated industry. The question of whether AI-generated misinformation about a drug constitutes a safety signal that should enter the pharmacovigilance workflow is one that medical safety teams have not yet definitively resolved. But the direction of travel is clear. As AI becomes a primary information channel for patients, the accuracy of AI-generated drug information becomes a pharmacovigilance variable.
The companies that treat AI monitoring as a safety function — not just a marketing or compliance function — will build the right internal ownership and resource allocation for the problem. The companies that treat it purely as a brand management issue will under-resource it and underestimate its urgency.
Building the Internal Case
Pharmaceutical regulatory and brand teams reading this face a practical challenge: how do you build an internal case for investment in AI drug information monitoring when the problem is new, the tools are emerging, and the ROI case is not yet written with historical data?
The strongest argument is regulatory liability prevention. One significant enforcement action, or one DOJ investigation in which the AI information environment around a drug is cited as part of a broader off-label promotion pattern, would cost orders of magnitude more than a multi-year AI monitoring program. This is an insurance argument, and it is the same argument that justified significant investment in social media compliance monitoring after the first FDA warning letters about pharmaceutical social media in 2010 and 2011.
The second argument is competitive intelligence. If your brand team could know, with systematic regularity, that your primary competitor’s AI profile in a key therapeutic category is inaccurate in ways that favor your drug’s clinical differentiation, that is actionable intelligence. The reverse is equally true: knowing that your drug’s AI profile is inaccurate in ways that favor your competitor gives you a problem to solve.
The third argument is patient support efficiency. If AI systems are consistently generating queries from patients about a specific dosing misunderstanding, that is signal about a patient education gap that your patient support program should address. The monitoring investment generates insights that improve the effectiveness of downstream commercial investments.
Key Takeaways
AI is now a primary drug information channel. Patients and clinicians query AI assistants before and sometimes instead of consulting prescribing information or healthcare professionals. What those AI systems say about your drug shapes perception, decision-making, and behavior.
The gap between AI responses and approved labeling is real and measurable. Systematic auditing of major AI platforms reveals consistent discrepancies from approved drug labels — in indication framing, dosing characterization, safety profile description, and competitive positioning.
Regulatory risk is not confined to what manufacturers publish. The AI information environment around a drug can create regulatory exposure even when the manufacturer has not created that content. Off-label descriptions, outdated safety profiles, and competitive conflation are AI-mediated risks that regulatory affairs teams need to map.
Traditional monitoring tools do not cover this channel. Social listening, media monitoring, and adverse event mining were not built to capture AI-generated drug information. Platforms like DrugChatter, purpose-built for AI drug information monitoring, fill a gap that existing tool stacks cannot.
Patient safety is the core issue. AI dosing errors, contraindication omissions, and indication drift are not marketing problems. They are safety problems with marketing and regulatory consequences. Internal ownership and resource allocation should reflect that priority.
The time to build monitoring capability is now, not after an incident. The companies building AI monitoring workflows in 2025 will have historical baseline data, established methodology, and practiced regulatory response protocols when the first enforcement action or significant adverse event involving AI-sourced drug information arrives.
FAQ
Q: Is an AI chatbot legally considered a promotional channel under FDA regulations?
A: Not currently, and not straightforwardly. FDA’s existing guidance on internet and social media promotion applies to manufacturer-controlled communications. An independent AI system generating responses about a drug based on publicly available training data sits outside that framework. However, the legal analysis is not static. If an AI system incorporates content that a manufacturer contributed — say, website copy, press releases, or un-blinded clinical presentations — the manufacturer’s role in shaping the AI’s output becomes a relevant factor. Legal teams at several large pharmaceutical companies are currently developing position papers on this question, and FDA has not issued definitive guidance. The absence of clear rules makes proactive monitoring more important, not less: you need to know what AI is saying before regulators ask you whether you knew.
Q: How often do major AI models update their drug information, and how should that affect monitoring cadence?
A: The update cadence varies significantly by platform and model architecture. Retrieval-augmented AI systems — those that retrieve information from live web sources at query time — can reflect new information within days of publication. Models that rely primarily on static training data may not reflect significant label updates for months, or at all if the cutoff predates the change. This creates a two-tier monitoring need. For drugs with recent label updates, active monitoring of AI responses for reflection of the update should begin immediately after the label change is effective. For stable products, quarterly auditing may be adequate. For drugs in high-activity therapeutic areas — oncology, rare disease, immunology — where the surrounding literature evolves rapidly, monthly monitoring better captures the signal.
Q: Can pharmaceutical companies submit corrective information to AI platforms when inaccuracies are discovered?
A: Formally, most major AI platforms have feedback mechanisms, and several have begun establishing healthcare-specific content partnerships. OpenAI, Google, and Microsoft have signaled openness to partnerships with authoritative health information sources. The practical efficacy of these channels varies considerably. Reporting an inaccuracy through a general feedback interface rarely results in traceable model correction. More structured approaches — establishing formal data-sharing agreements with AI platform operators, ensuring prescribing information appears in authoritative sources that AI retrieval systems preferentially index, and engaging with AI platforms’ healthcare advisory programs — are more likely to have measurable impact. This is an evolving space, and the pharmaceutical industry’s collective approach will matter: coordinated engagement from multiple pharmaceutical companies carries more weight than individual brand team requests.
Q: What is the relationship between AI drug information accuracy and patient support call center volume?
A: The data on this is preliminary but directionally consistent. Patient support teams at several large pharmaceutical companies have begun tracking whether callers report having received conflicting information from AI assistants. Where tracked, these instances correlate with call types that suggest informational confusion — questions about dosing that suggest the caller has received an incorrect number, questions about interactions that the approved label does not list as contraindicated, or questions about off-label uses that the patient encountered in an AI-generated response. As AI use increases among patients managing chronic conditions, the intersection with patient support will likely grow. From a commercial standpoint, this means AI monitoring insights can directly improve patient support efficiency by identifying and correcting the specific informational mismatches that are generating unnecessary calls.
Q: What role does scientific publication strategy play in shaping AI drug profiles?
A: Significant and underappreciated. AI models trained on biomedical literature weight published peer-reviewed content heavily. A drug’s representation in AI responses reflects, to a meaningful degree, the volume, recency, and framing of its peer-reviewed literature. A drug with a strong publication strategy — frequent, high-quality journal publications, robust presence in systematic reviews, consistent representation in guidelines documents — will tend to have more accurate and more favorable AI representation than a drug that is discussed primarily in conference abstracts, press releases, or online forums. This means that scientific publication strategy, traditionally managed as a medical affairs and regulatory function, has a direct bearing on the AI information environment and should be coordinated with whatever AI monitoring program the company implements. Knowing where the AI profile is inaccurate or unfavorable helps prioritize where publication gaps need to be closed.
Get sharp analysis on AI-generated drug information, off-label curiosity, pharmacovigilance blind spots, and the shifting intersection of pharma, search, and large language models. No fluff. Just the signals that matter.