The standard pharmacovigilance process has a timing problem
Adverse events travel faster than case reports. A patient posts about a rash on Reddit on Tuesday. Their physician files a report on Friday. A regulatory submission might arrive three weeks later. By then, dozens more patients have posted similar experiences, compared notes, and in some cases switched drugs entirely.
Drug companies have spent billions building structured reporting pipelines, and those pipelines are not going away. But the gap between when signals emerge and when safety teams see them has quietly become one of the most exposed operational risks in the industry. Artificial intelligence is closing that gap in ways that traditional pharmacovigilance infrastructure cannot.
This piece examines how AI monitoring works across the pharmacovigilance function — from unstructured adverse event detection to brand share measurement — and what it means for drug companies trying to stay ahead of regulatory risk rather than respond to it.
The Real-Time Signal Problem in Drug Safety
Pharmacovigilance teams operate under a regulatory framework designed for structured data: individual case safety reports (ICSRs), periodic safety update reports (PSURs), and risk management plans. These systems exist for good reasons. They enforce consistency, chain of custody, and medical review. But they were built before patients became publishers.
Today, roughly 80% of online health conversations happen in spaces that traditional drug safety systems never touch: Reddit threads, Facebook groups, disease-specific forums, app store reviews, and consumer health platforms. Patients describe drug experiences in plain language — ‘this medication made my heart race at 3 a.m.’ — without knowing the MedDRA term for palpitation or the concept of causality assessment. They are not trying to file adverse event reports. They are talking to each other.
The volume alone makes manual review impossible. A mid-sized pharmaceutical company with a portfolio of ten branded drugs might generate hundreds of thousands of social and forum mentions per month. A blockbuster oncology drug in active post-marketing surveillance can generate that in a week. No safety team, regardless of size, reads all of it. <blockquote> ‘Between 2010 and 2023, user-generated health content on public online platforms grew at a compound annual rate exceeding 22%, outpacing the growth of formally reported adverse events by a factor of roughly six to one.’ — IQVIA Institute for Human Data Science, 2023 Digital Health Report </blockquote>
The result is a structural asymmetry: patient signal is abundant and accelerating, while the infrastructure that captures it lags. AI monitoring does not replace the structured ICSR process. It provides the upstream layer that tells safety teams where to look and how urgently.
What AI Actually Does in a Pharmacovigilance Context
The term ‘AI monitoring’ covers several distinct capabilities that are often conflated. It helps to separate them.
Natural language processing for adverse event detection is the foundational layer. NLP models scan unstructured text — posts, reviews, transcripts, clinical notes — and identify mentions that have the structure of an adverse event: a drug name, a patient (or patient-adjacent) subject, and a reported experience that might constitute a health effect. Identifying all three in informal language requires models trained on both medical vocabulary and colloquial patient speech. ‘Made me feel like my heart was going to explode’ and ‘experienced cardiac palpitations’ describe the same phenomenon. A general-purpose language model handles neither well; a domain-adapted one handles both.
Entity recognition and normalization maps what patients say to what regulators need. When a patient writes ‘my fingers went numb,’ the NLP layer needs to recognize numbness, identify it as a potential neurological symptom, map it to the appropriate MedDRA preferred term (peripheral sensory neuropathy or paraesthesia, depending on context), and flag it with the drug name mentioned in the same post or thread. This is not a dictionary lookup problem. It requires understanding context, negation (‘I was worried about numbness but never experienced it’), speculation (‘I wonder if this drug causes memory problems’), and temporal reference (‘had the worst headache of my life last week’).
Causality and severity estimation gives safety teams a prioritization layer. Not all adverse event signals warrant equal attention. An AI system that surfaces 10,000 potential cases without ranking them has created a different kind of noise problem. Modern pharmacovigilance AI assigns preliminary causality scores — assessing how likely the drug is to have caused the reported event based on temporal relationship, biological plausibility, and language cues — and severity flags based on whether the reported experience maps to serious adverse event categories under ICH E2A guidelines.
Off-label use detection is one of the most commercially and regulatorily sensitive capabilities. When patients discuss a drug for an indication it is not approved for, that information carries dual implications: a potential safety signal in a population the drug was not studied in, and a regulatory risk if the company appears to be aware of and not acting on off-label use. AI systems that monitor for off-label patterns give safety and legal teams the early visibility they need to respond appropriately.
Brand share and sentiment tracking sits alongside safety monitoring but serves a different internal customer. Commercial and market access teams want to know how patients and physicians talk about their drug relative to competitors, whether switching behavior is driven by side effect perception or efficacy, and what specific experiences are driving prescriber hesitation. This is voice-of-customer work, but it happens in the same data streams and often with the same underlying models as adverse event detection.
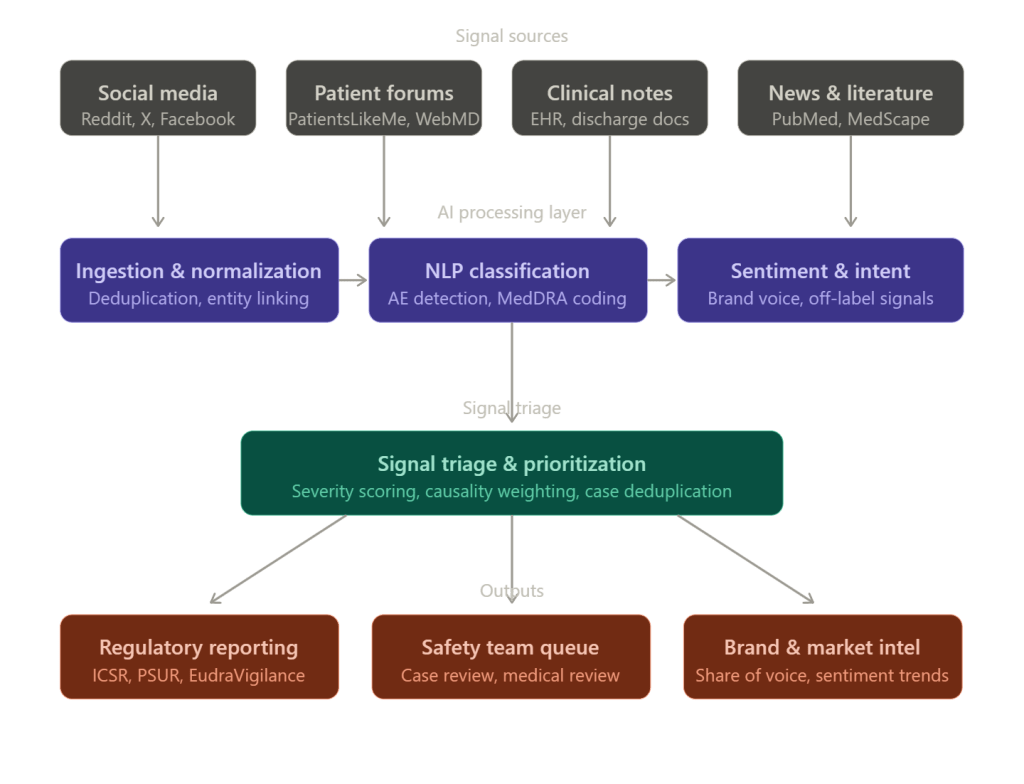
How the Monitoring Architecture Works
A well-built pharmacovigilance AI monitoring system has five operational layers, and understanding each helps drug companies evaluate vendor offerings or internal build-versus-buy decisions.
Data ingestion is where most systems either succeed or fail quietly. The sources are heterogeneous: public social platforms with rate-limited APIs, subscription-based medical databases, proprietary EHR feeds, call center transcripts, and field force contact records. A monitoring system that covers Reddit but misses condition-specific forums like CancerCompass or PatientsLikeMe has blind spots that can be significant for specific therapeutic areas. The ingestion layer also needs to handle multilingual content — a German Alzheimer’s patient group and a Brazilian diabetes forum contain patient experience data as relevant as their English-language equivalents, and the regulatory obligation to capture adverse events does not stop at a country’s border.
Deduplication and cross-source linking prevents the same patient experience from being counted multiple times. A patient who posts on Reddit, shares the same story in a Facebook group, and mentions it in an app review has described one potential adverse event, not three. Without deduplication, safety teams overestimate signal prevalence and may over-report, creating regulatory noise. Modern systems use embedding-based similarity matching and user pattern recognition to identify probable cross-source duplicates before a case reaches human review.
NLP and classification is the core intelligence layer. This is where drug mentions are extracted, adverse events are identified, MedDRA coding is proposed, and severity flags are applied. The performance metric that matters here is recall — how many real adverse events does the model find — balanced against precision, how many of the flagged items are actually adverse events. A system with 95% precision and 60% recall misses four out of ten real signals. In pharmacovigilance, that is not a model accuracy problem. It is a patient safety problem.
Triage and human review integration is the handoff point between AI output and safety professional judgment. No regulatory agency currently accepts AI-generated ICSRs without human medical review, and there is no indication that will change in the near term. What AI does is compress the time and labor required to get to that review. A trained safety associate who would have spent four hours reviewing 200 social posts to find eight potential adverse events can instead review a pre-filtered queue of 12 AI-prioritized cases in 45 minutes. The AI does not replace the safety associate’s judgment. It allocates her time.
Downstream regulatory and commercial integration determines whether the monitoring system creates value or creates a separate data silo. Safety findings need to flow into existing case management systems (Argus, Veeva Vault Safety, ARISg). Brand intelligence needs to reach brand teams in formats they actually use — dashboards, not CSV exports. Systems that require safety teams to manually re-enter AI-detected cases into their case management platform add process friction that erodes adoption.
Regulatory Expectations: What the FDA and EMA Have Said
The regulatory framework around AI-assisted pharmacovigilance is evolving, but it is not a blank slate.
The FDA’s 2021 guidance on digital health technologies and the 2023 discussion paper on AI in drug development both establish that the agency expects sponsors to understand and document the limitations of any AI tool used in regulatory-adjacent processes. For pharmacovigilance specifically, the FDA has indicated through public statements at the Drug Information Association (DIA) annual meetings that it expects companies using AI for signal detection to maintain auditable records of what the system surfaced, how it was prioritized, and what disposition the safety team applied.
The EMA’s pharmacovigilance risk assessment committee (PRAC) published a reflection paper on the use of artificial intelligence in pharmacovigilance in 2023, noting that AI tools can be used to support signal detection and case processing but must not reduce the quality or completeness of individual case safety reports submitted through EudraVigilance. PRAC’s position is effectively that AI is acceptable as a detection and efficiency tool, not as a replacement for medical judgment in causality assessment.
What both agencies have not done is prohibit AI monitoring. What they have done is make clear that companies using it need to be able to explain what they did and why. That requirement for explainability has direct implications for tool selection: black-box models that produce outputs without auditable reasoning paths create compliance exposure.
Drug companies operating in Japan face an additional consideration. PMDA (Pharmaceuticals and Medical Devices Agency) has been more conservative than FDA or EMA in accepting AI-derived evidence in regulatory submissions, though it has begun engaging with AI in clinical trial contexts. For pharmacovigilance, the practical implication is that Japanese language monitoring requires validated NLP models, not simple translations of English-trained models.
The Specific Risk of Not Monitoring AI Conversations About Drugs
There is a dimension of AI monitoring that pharmacovigilance teams are only beginning to account for: what AI systems themselves are saying about drugs.
Large language models — Claude, GPT-4, Gemini, and others — are now regularly consulted by patients for medical information. A patient managing a chronic condition who asks an LLM ‘what are the side effects of [drug]’ receives a response synthesized from the model’s training data, which may include outdated package insert information, user-generated forum posts from before the model’s training cutoff, and published literature that predates label changes. If the model’s output is inconsistent with the current approved label — for example, if it omits a recently added black box warning — that creates both a patient safety concern and a regulatory information environment that is working against the manufacturer’s communications.
Companies like DrugChatter have built monitoring capabilities specifically for this category of risk: tracking what AI systems say about specific drugs, identifying discrepancies with current approved labeling, and giving pharmacovigilance and regulatory affairs teams visibility into AI-generated medical information before it shapes patient behavior. This is not a theoretical risk. Patients who ask an LLM about drug interactions and receive an incomplete or outdated answer may make dosing decisions based on that information without consulting a physician.
The monitoring challenge here is different from social media surveillance. Social media involves users generating content. LLM monitoring involves tracking the outputs of probabilistic systems that give different responses to similar queries, that change behavior with model updates, and that are used by millions of patients who never label themselves as drug users in any database.
For regulatory affairs teams, the concern is how to respond when an AI system consistently understates a known risk. There is no established regulatory pathway for correcting an LLM’s output the way there is for correcting a misleading journal advertisement. But regulators are paying attention: the FDA’s Emerging Technology Program has begun informal discussions with sponsors about AI-generated health information, and it is reasonable to expect formal guidance within the next two years.
Brand Share Monitoring: The Commercial Case for the Same Infrastructure
The data infrastructure built for adverse event detection has a direct secondary use in commercial intelligence, and drug companies that treat these as separate investments are leaving money on the table.
Brand share of voice — the proportion of online drug-related conversations that mention a given brand versus its competitors — was traditionally measured through market research surveys and pharmacy data. Both methods are retrospective and episodic. Social and digital monitoring gives commercial teams a real-time signal that neither can match.
The practical application is straightforward. A brand team managing a diabetes drug wants to know, week by week, whether patients are discussing their drug or a competitor’s in the context of weight loss side effects, injection site convenience, or cardiovascular outcomes. They want to know whether prescriber commentary on LinkedIn or Doceree skews positive or negative relative to the same competitor. They want to understand whether a competitor’s new indication approval is shifting the conversation in ways that affect their drug’s perceived positioning.
This is the same underlying data as pharmacovigilance monitoring — drug mentions in public digital channels — but the extraction and analysis targets different features. The pharmacovigilance layer asks ‘is this a potential adverse event?’ The commercial layer asks ‘what does this tell us about brand perception?’
The operational efficiency case for integrating both on a single monitoring platform is significant. Running two separate vendor contracts for social listening — one for safety, one for commercial — produces overlapping data acquisition costs, inconsistent methodology, and organizational friction when the safety team’s findings have commercial implications (or vice versa). A single platform with separate output pipelines for safety and commercial teams eliminates that redundancy.
There are compliance guardrails that must be preserved. Commercial teams cannot access individual case information from the pharmacovigilance monitoring stream. The data separation between adverse event reporting and marketing intelligence must be maintained to avoid regulatory questions about promotional intent. But at the aggregate analytics level — brand share percentages, sentiment trends, topic frequency distributions — there is no regulatory restriction on commercial access, and the insights are directly actionable.
The MedDRA Coding Challenge and Why It Matters
Accurate MedDRA coding is the translation layer between patient language and regulatory language, and it is where most pharmacovigilance AI systems earn or lose their credibility with safety teams.
The MedDRA hierarchy has approximately 26,000 lowest-level terms, organized into preferred terms, high-level terms, high-level group terms, and system organ classes. An AI system that maps ‘stomach pain’ to ‘abdominal pain NOS’ rather than distinguishing between ‘upper abdominal pain’ and ‘lower abdominal pain’ may be producing technically valid codings, but it is losing clinical information that affects case assessment and signal detection analysis.
The practical consequence is that poorly coded AI-detected cases create more rework for medical reviewers, not less. If a safety associate receives a case coded to a generic preferred term and needs to re-evaluate the source text to apply a more specific code, the efficiency gain from AI detection has been partially or fully offset.
Drug companies evaluating pharmacovigilance AI vendors should request evidence of MedDRA coding accuracy, specifically precision and recall at the preferred term level on a holdout set that includes the types of informal patient language typical of social media, not just clinical trial narratives. The gap between performance on structured clinical text and performance on Reddit posts is larger than most vendors disclose.
Implementation Considerations: What Actually Goes Wrong
The pharmacovigilance AI monitoring implementations that fail tend to fail in predictable ways. Understanding these failure modes helps drug companies avoid them.
Scope creep in data sources is the most common operational failure. A system designed to monitor ten social platforms is expanded to twenty over eighteen months without corresponding validation. The additional sources may use different languages, different conversation norms, or different user demographics that the model was not trained on. Performance degrades, safety teams notice a higher false positive rate, and trust in the system erodes.
Alert fatigue follows directly from uncalibrated models. A pharmacovigilance AI system that surfaces 500 potential adverse events per week when the safety team can meaningfully review 80 has not improved the process. It has replaced one kind of backlog with another. The calibration of model thresholds to match the capacity and risk tolerance of the safety organization is not a one-time configuration task. It requires ongoing dialogue between data scientists and pharmacovigilance professionals.
Regulatory documentation gaps emerge when companies deploy AI monitoring without establishing the corresponding audit trail infrastructure. If the FDA asks how a safety signal was detected, the company needs to be able to show the source post, the AI output, the human review decision, and the disposition. Systems that do not log this chain of events create retroactive documentation problems that are expensive and time-consuming to resolve.
Organizational ownership disputes are underappreciated. Pharmacovigilance AI monitoring sits at the intersection of safety, regulatory affairs, commercial, and IT. When it is owned exclusively by safety, commercial intelligence use cases languish. When IT owns it, clinical validity suffers. When no one owns it clearly, it becomes a shared resource that nobody maintains well. The drug companies with the most effective implementations have appointed a cross-functional governance body with a safety professional as the final decision authority on data use and case handling.
Measuring ROI: The Numbers That Make the Case Internally
Drug companies getting budget approval for pharmacovigilance AI monitoring need to translate operational benefits into financial terms. The ROI case has four components.
Labor cost reduction in case processing is the most direct measure. If an AI triage layer reduces the time a safety associate spends per potential adverse event from 45 minutes (reading through forum threads manually) to 12 minutes (reviewing AI-pre-processed cases), the math is straightforward on a per-associate, per-year basis. For a company processing 50,000 potential cases annually, that efficiency gain represents several hundred thousand dollars in labor costs, depending on market and seniority level.
Earlier signal detection reducing regulatory action costs is harder to quantify but orders of magnitude larger in value. When a safety signal that would have taken six months to surface through spontaneous reporting is detected in week three via social monitoring, the company has the option to proactively engage with FDA rather than respond to a safety concern the agency has already identified. The cost difference between voluntary label update and a complete response letter from FDA requesting additional safety data can run into tens of millions of dollars in direct costs and market impact.
Reduced duplicate case submission from deduplication capabilities has a measurable compliance value. Over-reporting to regulatory databases creates noise for regulators and can trigger unnecessary safety inquiries. Under-reporting creates compliance exposure. Accurate deduplication improves both.
Commercial intelligence value is the component that often tips budget decisions when safety ROI alone is insufficient. Quantifying the value of knowing six weeks earlier than a competitor that a side effect narrative is driving prescriber switching — and being able to respond with targeted medical education — requires judgment about market share stakes. For drugs in competitive therapeutic categories, that judgment usually yields a large number.
The Case for Continuous vs. Periodic Monitoring
Many drug companies currently operate pharmacovigilance social monitoring on a periodic basis: weekly or monthly sweeps through digital channels. This approach made sense when the monitoring was done manually. It makes less sense when AI handles the ingestion.
The case for continuous monitoring is not about finding more adverse events. It is about finding them at the right time. Drug safety signals do not follow a weekly reporting schedule. They emerge when patients take a drug, experience something, and decide to talk about it. A patient who posts about a serious adverse event on a Sunday evening is not well-served by a monitoring system that next runs on Friday.
Continuous monitoring does not require continuous human review. The model can run continuously, building a triage queue that safety associates review on their normal schedule. What continuous monitoring provides is that no signal sits undetected for a week because it happened to occur between monitoring cycles. For serious adverse events — the ones with potential regulatory and patient safety implications — that timing difference matters.
The infrastructure cost difference between batch-periodic and continuous monitoring is smaller than most IT teams estimate, particularly for companies already paying for cloud-based NLP processing on a consumption model. The organizational resistance to continuous monitoring usually comes from safety teams worried about being overwhelmed by a continuously updating queue, which is a calibration problem, not an architecture problem.
What to Look For in Pharmacovigilance AI Vendors
The pharmacovigilance AI monitoring vendor market has expanded significantly since 2020. Evaluating vendors requires clarity about what good looks like.
Domain-adapted models trained specifically on pharmacovigilance use cases outperform general-purpose language models on the specific task of adverse event detection in patient language. Ask vendors for published or auditable evidence of their model’s performance on informal patient text, not just on structured clinical narratives.
MedDRA coding accuracy at the preferred term level should be provided with precision and recall data, not just general accuracy figures. Vendors who can only provide accuracy — which averages precision and recall — may be hiding a recall problem that matters more than precision in safety contexts.
Regulatory audit trail documentation should be a standard feature, not a custom implementation request. If a vendor cannot show you an audit trail of how a specific case was detected, scored, and dispositioned, they have not built for the regulatory environment that their customers operate in.
Multilingual capability with validated models matters for any company with products approved in more than two markets. Translation-based approaches — running content through machine translation before applying an English-language model — introduce errors that compound. Native multilingual models perform meaningfully better on safety-relevant terms.
Integration with existing case management systems determines whether the tool reduces or adds to process complexity. Ask for evidence of live integrations with Argus, Veeva Vault Safety, or the case management system your organization uses. A proof-of-concept API connection is not the same as a production-validated integration.
Platforms like DrugChatter address the specific question of what AI systems say about drugs, which is a monitoring category that most traditional pharmacovigilance vendors do not cover. As LLMs become more embedded in how patients find health information, this category of monitoring will become a standard expectation rather than an early-adopter capability.
Building Internal Competency vs. Buying External Capability
Drug companies reliably underestimate what it takes to build pharmacovigilance AI monitoring internally and reliably underestimate how much control they lose when they outsource it entirely.
Building internally gives full control over model training data, audit trail design, and data governance. It allows the safety team to tune the model against the company’s specific drug portfolio, therapeutic areas, and patient populations without waiting for a vendor release cycle. The cost is significant: model development, validation, ongoing maintenance, and the data science talent to do all of it.
Buying externally provides faster deployment, vendor-maintained validation documentation, and access to models trained on larger datasets than any individual company could assemble. The cost is dependence on vendor roadmaps, limited insight into model internals, and the need to trust vendor claims about performance.
The most defensible approach for most drug companies is a hybrid: a vendor platform for broad digital channel monitoring, with internal capability to run secondary analysis on cases that reach the case management system. This preserves the efficiency gains of vendor-built ingestion and NLP while keeping regulatory-sensitive analysis under the company’s direct control.
Key Takeaways
Drug companies that treat pharmacovigilance AI monitoring as an IT project rather than a safety and compliance imperative will find themselves revisiting the decision after a regulatory inquiry makes the case for them.
The core operational value is time. AI monitoring compresses the gap between when adverse events emerge in patient conversations and when safety teams see them. For serious adverse events, that gap currently runs from weeks to months. Closing it to days changes the regulatory dynamic from reactive to proactive.
MedDRA coding quality, model recall performance on informal patient language, and audit trail completeness are the three metrics that determine whether a pharmacovigilance AI system is operationally useful or operationally performative. Vendors who cannot provide clear evidence on all three should not be in the final evaluation.
The same data infrastructure that serves pharmacovigilance safety monitoring serves commercial intelligence on brand share and patient sentiment. Running separate systems for both functions doubles cost and creates methodological inconsistency. Integrated platforms with separate output pipelines for safety and commercial users are the right architecture.
LLM monitoring — tracking what AI systems say about specific drugs — is an emerging category that few safety teams have formalized. Regulatory frameworks for it do not yet exist, but the underlying risk does. Drug companies that begin monitoring AI-generated drug information now will be ahead of the regulatory curve when guidance arrives.
Continuous monitoring is technically straightforward and organizationally underutilized. The resistance to it is usually about queue management, which is a calibration challenge, not a fundamental constraint.
FAQ
Q: Does AI-detected adverse event content from social media need to be reported to the FDA as an ICSR?
Not automatically. The FDA’s regulations on expedited reporting (21 CFR 314.81 and 21 CFR 312.32) apply to adverse events that are both serious and unexpected, where the company has certain knowledge of the event. Social media posts represent solicited or unsolicited reports, and the FDA’s 2013 guidance on social media and internet communications clarifies that companies are responsible for reporting adverse events from sources they actively monitor. If your company has a system monitoring social media for adverse events — which AI makes practical — posts meeting the ICSR threshold are reportable. Companies should have their regulatory affairs counsel review the specific operationalization of ‘actively monitor’ in their context, since the monitoring infrastructure itself creates the reporting obligation.
Q: How do pharmacovigilance AI systems handle negation and speculation in patient posts?
This is one of the harder technical problems in medical NLP. Negation (‘I was worried about hair loss but didn’t experience it’) and speculation (‘I wonder if this is causing my fatigue’) must be distinguished from positive adverse event reports to avoid false positives. Domain-adapted models trained on pharmacovigilance text handle this significantly better than general-purpose models because they have been trained on examples of how patients express uncertainty about drug effects. Standard performance benchmarks for medical NLP — such as the n2c2 shared tasks — include negation detection as a separate evaluation dimension, and vendors should be able to provide negation-specific performance metrics.
Q: What is the regulatory status of AI-generated MedDRA coding?
AI-proposed MedDRA coding requires human medical review before appearing in a regulatory submission. Neither the FDA nor EMA currently accepts AI-coded cases without human validation. The operational question is whether AI coding proposals — even when subject to review — introduce systematic bias that shifts the distribution of coded terms in ways that affect signal detection analyses. Companies with large AI-processed case volumes should periodically audit their coded case distributions against their pre-AI baseline and against industry benchmarks to identify any systematic drift.
Q: How should a drug company respond if an LLM is consistently misrepresenting a drug’s safety profile?
There is no established regulatory mechanism for directly correcting LLM output, but several response options exist. Companies can engage with the AI company’s trust and safety or policy teams to flag factually incorrect health information — most major LLM developers have processes for this. Companies can also increase the volume and accessibility of accurate label information in the digital commons (company websites, medical affairs publications, patient information resources) to improve the signal quality available to LLMs in future training. Working with medical communication teams on platform-specific LLM grounding is an emerging practice. Regulatory affairs should document the discrepancy and the company’s response in case the FDA’s Emerging Technology Program or similar bodies request information.
Q: Can field medical affairs teams use AI monitoring outputs for medical education planning?
Yes, with appropriate governance controls. Aggregate insights from AI monitoring — which adverse events are most commonly discussed, which clinical questions physicians are asking on professional platforms, which patient populations are expressing the most confusion about dosing — are directly useful for medical affairs planning of publication strategy, advisory board agenda, and medical science liaison (MSL) focus areas. The control that must be maintained is that individual case information from the pharmacovigilance monitoring stream is not shared with commercial-facing functions, including medical affairs teams in contexts where they are supporting promotional activities. Aggregate, de-identified trend data does not carry the same restriction.





