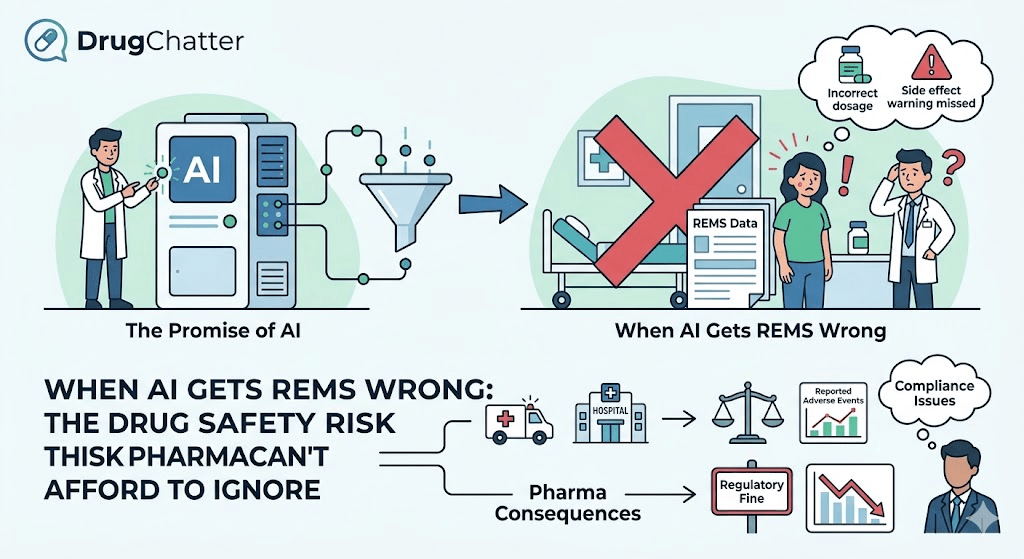
A physician opens ChatGPT and types: ‘What are the prescribing requirements for isotretinoin?’ The model replies with a confident, readable summary. It mentions severe acne, birth defect risk, contraception, pregnancy testing. It does not mention iPLEDGE. It does not explain that the prescriber must be enrolled in a federal risk management program before a single prescription can be written. It does not say that the pharmacy must be certified, that the patient must complete a monthly attestation, or that isotretinoin cannot legally be dispensed without a confirmed negative pregnancy test tied to a specific window.
The physician gets a plausible-sounding answer that omits the mechanism designed to keep the drug safe. That omission is not a minor editorial lapse. It is a failure of the most consequential element of isotretinoin’s regulatory architecture.
This is the core problem with AI and REMS — and it is happening right now, at scale, across ChatGPT, Gemini, Claude, Perplexity, and Microsoft Copilot, every time a prescriber, patient, pharmacist, or caregiver asks a question that touches a drug with a Risk Evaluation and Mitigation Strategy.
What Is a REMS Program and Why Does Accuracy Matter So Much?
A Risk Evaluation and Mitigation Strategy is a legally mandated safety program that the FDA can require when a drug’s risks are serious enough that standard labeling alone cannot manage them. The authority comes from Section 505-1 of the Federal Food, Drug, and Cosmetic Act, enacted under the FDA Amendments Act of 2007.
REMS programs vary considerably in their requirements. The simplest involve only a Medication Guide — a structured patient handout. The most complex include Elements to Assure Safe Use, known as ETASU, which can mandate prescriber certification, pharmacy enrollment, patient registration, laboratory monitoring at defined intervals, restricted distribution channels, and in some cases, the physical location where a drug can be dispensed.
As of early 2025, DrugChatter and other industry monitors have noted approximately 73 active REMS programs in the United States, with roughly 95% of them incorporating at least some ETASU requirements. The drugs covered span oncology, psychiatry, reproductive medicine, pain management, and immunology. They include some of the most commercially significant and clinically complex products on the market.
Which Drugs Carry REMS Requirements That AI Routinely Gets Wrong?
The programs with the highest error risk in AI outputs are the ones with multi-step, stakeholder-spanning requirements that cannot be reduced to a single rule. Isotretinoin’s iPLEDGE program tops the list. The program mandates a computer-based system with verifiable, traceable links among prescriber, patient, pharmacy, and wholesaler. Prescribers must be enrolled. Pharmacies must be certified. Patients must be registered, must demonstrate understanding of the risk through a comprehension questionnaire, and must produce a negative pregnancy test within a specific window before each monthly prescription. The window is not flexible.
The Clozapine REMS — prior to its removal effective June 13, 2025 — required absolute neutrophil count monitoring at defined intervals, with prescribing and dispensing locked out if monitoring was not current. That program ran for years as one of the most restrictive distribution systems in U.S. pharmaceutical history, and AI models consistently described it incompletely or incorrectly even while it was active.
The Opioid Analgesic REMS, modified as recently as October 2024, covers extended-release and long-acting opioid analgesics dispensed in outpatient settings. The TIRF REMS Access program covers transmucosal immediate-release fentanyl products — drugs with significant abuse potential and narrow therapeutic windows. CAR T-cell therapies, expanded under the REMS framework in June 2024 to cover all six approved products, carry requirements around cytokine release syndrome and neurological toxicity management that changed substantially with that update.
Mifepristone, which has carried a REMS since 2000 and has seen its requirements modified repeatedly, including the elimination of the in-person dispensing requirement that allowed certified pharmacies to dispense directly with a prescription, sits at the intersection of clinical complexity and political salience. AI models asked about mifepristone’s dispensing requirements frequently reflect outdated information, often describing requirements that no longer apply or missing modifications that do.
How REMS Information Becomes Distorted in AI Training Data
The mechanism behind AI REMS errors is not mysterious. Large language models are trained on a snapshot of text collected before a cutoff date. REMS requirements change — sometimes substantially — after that date. The iPLEDGE program changed its requirements in February 2026, allowing prescribers to permit home pregnancy testing under certain conditions. Any model trained before that change will describe requirements that are no longer fully accurate.
The problem goes beyond knowledge cutoffs. REMS documents are regulatory instruments. They are not written for consumer consumption, and the secondary coverage of REMS programs — the news articles, patient forum posts, health website summaries, and blog content that AI models absorb in training — tends to oversimplify, omit steps, or describe the spirit of the program without the operational specifics that make compliance real. The model learns from that secondary coverage. The gap between ‘you need a pregnancy test’ and ‘you need a negative pregnancy test confirmed through the iPLEDGE portal within a defined window, completed by a certified prescriber, with a dispensing authorization that expires in seven days’ is the gap between general awareness and actual compliance.
‘In a sample audit of twenty branded drugs conducted by DrugChatter in 2024, 65% had at least one material factual error in AI-generated descriptions across the four major platforms — wrong dosing information, incorrect indication language, outdated contraindication lists, or mislabeled routes of administration.’
— DrugChatter / DrugPatentWatch audit data, 2024
REMS information is disproportionately likely to be wrong because it changes more frequently than most drug information, it is more complex than labeling summaries, and it is the category of information where omission carries the most direct patient risk.
Can AI Hallucinations About REMS Requirements Trigger FDA Regulatory Risk?
The short answer is yes — through two distinct pathways, and a third that is still forming.
The first pathway runs through a company’s own use of AI. In April 2026, the FDA issued its first warning letter explicitly citing artificial intelligence misuse in pharmaceutical manufacturing to Purolea Cosmetics Lab, a Michigan-based drug manufacturer. The company had used AI agents to generate drug product specifications, standard operating procedures, and master production and control records. When FDA investigators identified that the firm had not conducted process validation before distributing drug products, the owner explained that the AI agent had not told her that validation was required. FDA rejected that explanation. The agency made clear that companies using AI in document creation must review AI-generated outputs to confirm accuracy and actual CGMP compliance, and that the Quality Unit’s responsibility under 21 CFR 211.22(c) does not transfer to the AI system.
The holding extends beyond manufacturing. A pharma brand team that uses AI to draft REMS-related communications — patient materials, prescriber letters, pharmacy guidance — and fails to review those outputs for regulatory accuracy faces the same statutory risk. The AI’s failure to describe a requirement correctly does not excuse the sponsor’s failure to communicate it correctly. The sponsor remains responsible for the accuracy of materials that describe its REMS program, regardless of what tool was used to draft them.
Does the FDA Have Authority to Act on AI-Generated Drug Misinformation?
The FDA’s statutory authority under Section 301 of the FDCA prohibits false or misleading labeling, but that authority runs to labeling the manufacturer controls or disseminates. An AI platform generating inaccurate REMS descriptions is not currently treated as the manufacturer’s labeling, and no direct regulatory precedent as of mid-2026 holds a drug sponsor liable for AI outputs it did not produce or distribute.
That regulatory gap may not hold. The FDA has signaled expanding scrutiny of AI across regulated operations. Its January 2025 draft guidance — ‘Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products’ — establishes principles around AI validation and human oversight that are now being interpreted in enforcement contexts. The Purolea warning letter is, in the language of regulatory analysts, ‘an early signal.’ The FDA is watching how companies interact with AI, how they validate AI outputs, and whether they maintain genuine human oversight over AI-generated information in regulated settings.
What Happens When a Patient Skips a REMS Step Because of AI Advice?
A patient who reads an AI summary of their isotretinoin requirements and concludes they only need to ‘avoid pregnancy’ rather than enroll in iPLEDGE and complete monthly attestations has received information that creates real clinical risk. If they fill a prescription at a non-certified pharmacy — which an AI description omitting the certification requirement would not prevent — the pharmacist cannot legally dispense. In practice, that patient discovers the compliance requirement at the pharmacy counter, often after a delay that interrupts treatment.
The more serious scenarios involve REMS programs designed to prevent acute harm. A patient taking transmucosal fentanyl who asks an AI chatbot whether their dose can be adjusted between appointments and receives an answer that does not reflect the TIRF REMS Access program’s monitoring requirements and titration restrictions has received advice that contradicts a federally mandated risk management framework. Whether that advice causes harm depends on clinical context, but the regulatory machinery was built precisely to prevent that scenario.
How LLMs Describe REMS Drugs Differently From Each Other
ChatGPT, Gemini, Claude, Perplexity, and Microsoft Copilot do not produce identical outputs when asked the same REMS-related question. The divergence is not random — it reflects different training data weighting, different knowledge cutoffs, different retrieval strategies, and different levels of caution built into each system’s response patterns.
Which AI Platform Is Most Accurate on REMS Drug Information?
Systematic comparison across platforms reveals consistent patterns. Perplexity, which retrieves and cites live web sources, is more likely to surface current REMS documentation when that documentation is publicly indexed. Its outputs are also more verifiable — a user who sees a citation can follow the link and check. ChatGPT, particularly in versions without web search enabled, is most likely to describe REMS requirements as they existed during its training period, without flagging that requirements may have changed. Gemini draws heavily from Google’s search index and knowledge graph, which can surface current regulatory content but also reflects the full range of secondary coverage, including older or inaccurate summaries.
Claude tends toward epistemic caution — it is more likely to qualify its REMS-related answers and recommend verification with a healthcare provider or the FDA, but that caution does not prevent it from describing outdated requirements as current when it lacks up-to-date training data.
None of the major platforms performs reliably on the operational specifics of complex REMS programs. The difference between platforms is primarily in how they handle uncertainty, not in whether they get REMS details right.
Do LLMs Recommend Generic Drugs Over Branded REMS Products?
This question matters for pharma brand teams because REMS programs can cover all versions of a drug — brand and generic — or only the originator product. When AI models discuss drugs subject to REMS as generic alternatives, they may implicitly or explicitly suggest that the REMS requirements do not apply to the generic, or they may recommend the generic without clarifying that the same restrictions apply. For shared REMS programs like iPLEDGE, which covers all isotretinoin manufacturers, this creates misinformation about equivalence of access — a patient cannot bypass the REMS by switching from Absorica to a generic Claravis.
The competitive dimension compounds the problem. When a physician asks an AI tool to compare treatment options in a given therapeutic area, the AI’s framing of REMS requirements — as a burden associated with the branded product, as a complexity the generic avoids, or as a neutral feature of the drug class — directly shapes prescribing intent. Brand teams that are not monitoring these comparisons have no visibility into how their product is being positioned relative to competitors in the fastest-growing drug information channel in the world.
How Patients Ask About Drug Safety in AI Search — and What They Actually Receive
Patient queries to AI chatbots about REMS drugs follow predictable patterns. They ask about side effects, dosing, what to tell their doctor, whether they can drink alcohol, whether a medication is safe during pregnancy, and what happens if they miss a dose. These are not questions that directly reference REMS, but many of them touch REMS-regulated risks. An isotretinoin patient asking ‘Can I take isotretinoin if I’m not using birth control?’ is asking a question that the iPLEDGE program is specifically designed to answer through a structured enrollment and monitoring process. The AI answer that addresses the clinical risk without explaining the program’s requirements is factually incomplete in a way that matters.
Research published in preprint in early 2025 found that even LLMs developed specifically for medical purposes remain vulnerable to domain-specific hallucinations, often arising from reasoning failures rather than mere knowledge gaps. Patients without clinical training are less equipped to identify when an AI answer is incomplete or misleading, and they are more likely to act on it without verification.
Off-Label Discussions in AI: REMS Drugs and Unapproved Uses
REMS programs are built around specific approved indications. When AI models discuss off-label uses of REMS drugs, they are operating in territory where the risk management program’s requirements may not apply — but where the underlying safety risks that prompted the REMS almost certainly do.
How AI Models Discuss Off-Label REMS Drug Uses Without FDA Context
Isotretinoin is approved for severe nodular acne that has not responded to other therapies, including antibiotics. It is also used off-label for hidradenitis suppurativa, rosacea, and several other conditions. An AI model asked whether isotretinoin can be used for hidradenitis suppurativa will often describe the off-label use and its evidence base without noting that the teratogenic risk that makes iPLEDGE necessary applies regardless of indication. The REMS follows the drug, not the diagnosis.
Mifepristone has an approved indication for medical termination of intrauterine pregnancy, and it is used off-label in combination regimens for other conditions. The REMS — updated to allow certified pharmacy dispensing with a prescription — applies to the approved use. AI models that describe mifepristone’s pharmacology in the context of off-label uses frequently omit the REMS entirely, treating it as an indication-specific constraint rather than a drug-specific one.
For CAR T-cell therapies, where REMS requirements around cytokine release syndrome management apply regardless of the specific indication being treated, off-label use discussions in AI often strip the safety context entirely. A physician researching CAR T options for a patient with an off-label indication is operating in exactly the clinical setting where the REMS-mandated monitoring protocols are most important, and AI descriptions of those drugs frequently do not surface them.
Can AI Outputs Be Used for Pharmacovigilance? The REMS Signal Detection Problem
Pharmacovigilance teams at pharmaceutical companies are responsible for detecting, evaluating, and reporting adverse events associated with their products. Traditionally, that signal detection work draws on FDA Adverse Event Reporting System data, clinical trial safety databases, medical literature, and increasingly, social media monitoring. AI-generated content represents a new signal source — one that is both a channel for patient experience data and a potential source of misinformation that itself generates adverse outcomes.
The dual nature of AI as pharmacovigilance input creates a methodological challenge. If patients are asking AI chatbots about symptoms they are experiencing on a REMS drug, and the AI’s response influences whether they report those symptoms, continue their medication, or seek emergency care, then AI outputs become part of the causal chain for adverse event patterns. A pharmacovigilance team that monitors patient forums and Reddit but not AI chatbot responses is missing a rapidly growing segment of health information behavior.
Research published at arxiv in 2024 describing guardrails for LLMs in pharmacovigilance contexts explicitly flagged hallucination as a ‘pivotal concern’ in drug safety settings, noting that models ‘can generate fabricated information’ about adverse events and that ‘inaccuracies could lead to patient harm.’ The paper described mechanisms to detect incorrect drug names or adverse event terms — a recognition that the AI’s own errors are themselves a pharmacovigilance risk.
How Pharma Brand Teams Can Monitor AI Mentions of REMS Drugs
Pharmaceutical brand teams already invest substantially in monitoring what prescribers, patients, and payers say about their products. Digital shelf analytics, social listening, brand tracking surveys, and medical information call center data all feed into brand intelligence programs. AI monitoring is the gap most have not yet addressed.
What an AI Brand Monitoring Program for REMS Drugs Should Actually Include
Effective AI monitoring for a drug with REMS requirements needs to track four categories of output: factual accuracy against approved labeling, REMS-specific accuracy against current program documentation, competitive framing relative to other drugs in the therapeutic area, and patient-facing language and sentiment. These are distinct analytical problems that require different query batteries and different scoring frameworks.
Factual accuracy monitoring runs structured queries against current prescribing information and scores AI outputs for correctness. For a REMS drug, this layer must also include REMS-specific queries — questions about enrollment, monitoring requirements, certification, dispensing restrictions, and program logistics — scored against current program documentation, not labeling alone.
Competitive framing analysis monitors how AI models describe a drug relative to alternatives, whether REMS requirements are framed as burdens that competitors avoid, and whether the AI’s comparative responses favor or disadvantage the monitored brand. This analysis matters most when a drug’s REMS program is more restrictive than a competitor’s, or when generic alternatives are presented without equivalent safety framing.
Patient-facing language monitoring tracks how AI describes the drug in consumer-directed terms — what it says about convenience, side effects, and the practical experience of using a drug subject to REMS — and identifies emerging patient concerns before they consolidate into reputational issues or signal adverse event trends.
Tracking Share of Voice Across ChatGPT, Gemini, and Claude for REMS Drugs
AI share-of-voice for pharmaceuticals is not identical to traditional share-of-voice measurement. In traditional media, share of voice measures how much of the total conversation about a drug class a specific brand captures. In AI search, share of voice measures how often a specific brand is mentioned when a user asks a relevant question — and critically, where in the response it appears. First mention in an AI response carries different weight than a parenthetical mention in a list of alternatives.
The same query asked to ChatGPT, Gemini, Claude, and Perplexity can produce substantially different answers in terms of which drugs are mentioned first, what side effects are emphasized, and how comparative claims are framed. For REMS drugs, this platform-level variation matters because each platform reaches a different user population. Perplexity skews toward research-oriented users who follow citations. ChatGPT reaches the largest aggregate user base. Gemini captures users already in the Google ecosystem who may be asking health questions as part of a broader search session.
A monitoring program that queries only ChatGPT is missing the platform-specific variation that determines where a brand’s AI presence is strong, where it is weak, and where REMS-related misinformation is concentrated. DrugChatter runs structured query batteries across ChatGPT, Gemini, Claude, Copilot, and Perplexity simultaneously, logging outputs and scoring them for accuracy, regulatory alignment, sentiment, and competitive framing — giving pharmaceutical brand and medical affairs teams the equivalent of a digital shelf audit across the AI search channel.
How Often Should Pharma Companies Run AI Monitoring Queries on REMS Drugs?
For drugs with active REMS programs, weekly monitoring is the appropriate minimum cadence. Major AI platforms update their models on irregular schedules. A model update can shift outputs materially within days. A REMS program modification — like the iPLEDGE changes that took effect in February 2026, or the Opioid Analgesic REMS modification finalized in October 2024 — creates an immediate monitoring event. The AI models will not update their knowledge to reflect the change on the same schedule that the change takes effect. There is a lag, and the lag is unmeasured unless someone is monitoring.
Monthly monitoring is a minimum baseline for any commercially important brand. Quarterly monitoring — which is what most companies default to if they monitor at all — is too slow to detect changes in time to mount an effective response. When REMS documentation changes, the window for the AI’s training data to update is unpredictable. During that window, physicians and patients are receiving outdated information with the same confidence that the AI delivers current information. That window is not visible without active monitoring.
The Physician-Facing REMS AI Risk: What Prescribers Are Actually Asking
Physicians are using AI tools in clinical practice. The rate of adoption varies by specialty and setting, but surveys across 2024 and 2025 consistently find that a substantial share of practicing physicians have used ChatGPT, Gemini, or a clinical AI tool to look up drug information, dosing guidance, or drug interaction data. Many of those queries touch drugs with REMS requirements.
What Physicians Are Asking AI About Clozapine, Isotretinoin, and Opioid REMS Drugs
Physician queries about REMS drugs cluster around practical operational questions rather than clinical pharmacology. They ask how to enroll a patient in a REMS program, what the monitoring intervals are, whether a specific test result permits continued prescribing, how to handle a patient who misses a monitoring requirement, and what the dispensing restrictions mean for their specific practice setting. These are exactly the questions where REMS accuracy matters most — where an incomplete answer can delay care or, worse, enable a dispensing event that the REMS was designed to prevent.
For clozapine, the absolute neutrophil count monitoring requirement — which was enforced by the REMS system that locked prescribing and dispensing if monitoring was not current — required precise knowledge of the monitoring intervals and the consequence of non-compliance. An AI answer that describes ‘regular blood monitoring’ without specifying intervals and consequences gives a physician an impression of compliance without the information needed to achieve it.
The FDA removed the Clozapine REMS effective June 13, 2025, based on its re-evaluation and the advice of the November 2024 Joint Meeting of the Drug Safety and Risk Management Advisory Committee and the Psychopharmacologic Drugs Advisory Committee. AI models trained before that date — and some trained after it — continue to describe clozapine as requiring REMS enrollment. That is a different kind of error: not an omission of required steps, but a description of requirements that no longer legally exist. A physician or pharmacist who believes clozapine still requires REMS enrollment may create unnecessary administrative barriers to dispensing for patients who need it.
How AI Handles REMS Updates and Why Stale Information Is a Systemic Risk
REMS programs are not static. The FDA’s own REMS compliance program documentation describes REMS assessment as ‘an iterative and complicated process.’ Programs are modified as new data emerges, as burden assessments are completed, and as the FDA and sponsors agree that requirements can be loosened, tightened, or removed. Each modification creates a period during which AI models have outdated information and no mechanism to flag it as outdated.
The May 2024 FDA draft guidance introducing the REMS Logic Model — a structured framework for designing, implementing, and evaluating REMS programs — signals that the FDA views REMS as living programs that should be continuously evaluated and modified. That vision of dynamic, evidence-responsive REMS programs is in direct tension with how AI models handle program information: as static knowledge absorbed at training time, accurate or not, current or not, comprehensive or not.
Pharmaceutical companies whose REMS programs are modified face a specific communications risk. Their brand and medical affairs teams may update their own materials, retrain their field forces, and notify healthcare providers through direct communications. None of those actions changes what the AI models say. The AI channel is the gap that exists between the regulatory action and the information environment that physicians and patients actually encounter.
AI-Driven Patient Sentiment on REMS Drugs: What Forum Data and LLMs Reveal
Patient sentiment about REMS drugs is shaped not only by clinical experience but by how patients describe that experience to each other, to AI chatbots, and on platforms like Reddit, patient forums, and condition-specific communities. The REMS program itself generates patient sentiment — enrollment friction, monitoring burden, dispensing delays, and the perception of restriction all influence how patients talk about a drug and how they engage with it.
What Patients Say on Reddit About iPLEDGE and Other REMS Programs
Reddit’s r/Accutane community, with hundreds of thousands of members, contains extensive discussion of the iPLEDGE experience. The dominant patient sentiment around iPLEDGE is frustration with the monthly attestation window, confusion about the portal interface, and anxiety about dispensing delays when the process goes wrong. That patient-generated content feeds into the training data that shapes how AI models describe the iPLEDGE experience — which means that when AI describes the ‘requirements’ for isotretinoin, it is drawing on a mix of regulatory documentation, clinical summaries, and patient forum discussions that weight convenience and friction concerns alongside compliance requirements.
The resulting AI descriptions of iPLEDGE tend to convey the program’s complexity and burden accurately while underspecifying the specific operational steps. That pattern is directly traceable to the source material. Patients writing on Reddit are not listing enrollment steps; they are describing their experience. The AI learns from experience descriptions, not from procedural documentation.
Social listening tools that monitor Reddit, patient forums, and health communities give pharmaceutical companies visibility into the organic patient experience of their REMS programs. AI monitoring tools that query chatbots with patient-style questions give companies visibility into how the AI is translating and retransmitting that experience data back to future patients. Both channels are necessary. Neither alone is sufficient.
Can AI Sentiment Analysis Help Pharma Detect Emerging REMS Compliance Problems?
There is an emerging use case for AI-driven content analysis of patient communities to identify REMS compliance signals before they appear in FAERS data. When patients in online communities begin describing experiences that suggest they are not following monitoring requirements — either because they did not know the requirements or because they encountered barriers to compliance — that discussion pattern precedes the adverse event reports that would eventually surface through the formal pharmacovigilance system.
A pharmaceutical company running semantic analysis against iPLEDGE-related patient content could, in principle, detect increases in discussions about skipping pregnancy tests, accessing isotretinoin outside the certified pharmacy network, or misunderstanding the attestation requirements before those patterns manifest as clinical events. That is the pharmacovigilance value proposition of social listening applied to REMS monitoring — and it is largely unrealized by most sponsors.
The tool at DrugChatter bridges this gap by surfacing both what AI models are saying about a drug and what the underlying discourse that feeds those models looks like — giving brand and safety teams an integrated view of the AI information ecosystem around their products.
The Citation Problem: How AI Picks Its Sources for Drug Safety Information
When AI models describe REMS requirements, they are synthesizing information from the sources they were trained on — but the weight each source receives in that synthesis is not transparent. A model might weight a 2019 health website summary of the Clozapine REMS program alongside the FDA’s current REMS documentation, without the user knowing that the two sources describe different requirements or that one of them is now outdated.
Why AI Cites Patient Forums More Than FDA Guidance on REMS Programs
Volume, not authority, drives how much influence a source has on a language model’s outputs. There is vastly more patient-generated content about REMS drugs on the open internet than there is authoritative regulatory documentation. FDA REMS guidance documents are precise but sparse. Patient forums are imprecise but voluminous. Medical news coverage of REMS programs is more readable than the regulatory documents but often omits the operational specifics that matter for compliance.
The result is that AI models tend to describe REMS programs in the register of patient experience — they convey the ‘what’ of the program without the ‘how.’ They communicate that isotretinoin requires pregnancy prevention without specifying the portal, the window, the attestation, the certified pharmacy requirement, or the consequences of non-compliance. That is a description accurate enough to feel informative and incomplete enough to be operationally useless.
Perplexity’s web retrieval capability partially addresses this by pulling current FDA documentation when a query is phrased to trigger official source retrieval. But Perplexity is not the dominant AI tool in most clinical workflows, and the users most likely to ask REMS-relevant questions — patients navigating their treatment for the first time — are the least likely to phrase those questions in ways that trigger official source retrieval.
How Pharma Companies Can Influence What AI Cites About Their REMS Programs
This is the generative engine optimization question applied to pharmaceutical safety. AI citation behavior is not random. Models favor sources with high domain authority, clear factual statements, structured content that is machine-readable, and consistency with other high-authority sources. Pharmaceutical companies that make their REMS program documentation accessible, clearly structured, and consistently described across owned and earned channels are better positioned to have that documentation reflected in AI outputs than companies whose REMS information is buried in PDF documents on FDA.gov and not amplified anywhere else.
This is not a suggestion to game AI systems. It is a recognition that the information environment that shapes AI outputs is itself shapeable. A pharmaceutical company that publishes clear, structured, current REMS program guidance in machine-readable formats, that earns citations from high-authority health media, and that maintains consistent messaging across its digital owned properties is doing what the current information environment rewards. The alternative is allowing the AI’s training data mixture to determine how physicians and patients understand a federally mandated safety program — and that is a risk that can be managed.
What Eli Lilly, Novo Nordisk, and Other Pharma Leaders Should Learn From REMS AI Monitoring
The pharmaceutical companies best positioned to manage AI REMS risk are those that treat AI monitoring as an extension of their existing medical information and pharmacovigilance programs, rather than as a separate digital marketing function. The clinical stakes of REMS misinformation are too high to manage it as a brand awareness problem.
How AI Monitoring Fits Into a Pharma Company’s Medical Affairs Program
Medical affairs teams already own the clinical accuracy mandate for their companies’ drug information. They manage medical information hotlines, respond to unsolicited off-label inquiries, and govern the creation of medical education materials. AI monitoring belongs in that same governance structure. The questions that AI models are answering about REMS drugs are the same kinds of questions that medical information teams field through conventional channels — except that AI is fielding them at a scale and speed that no human team can match, without a quality control process, and without the institutional knowledge to know when it is wrong.
A medical affairs team that integrates AI monitoring into its quarterly safety review cycle can identify divergences between what AI models say about a drug and what the drug’s current labeling and REMS documentation require, then route those divergences to content strategy and regulatory affairs for assessment. The regulatory question — whether and how to respond to AI misinformation about a REMS program — is novel and unsettled, but the identification of the problem is straightforward once the monitoring infrastructure is in place.
Building an Internal AI REMS Monitoring Protocol: A Practical Framework
A functional AI REMS monitoring protocol for a drug under REMS has four components. The first is a query library — a structured set of questions that cover prescriber operational needs, patient experience questions, clinical pharmacology questions, and competitive framing queries. The library should be built from the actual question patterns that the medical information team encounters, supplemented with patient forum analysis to identify the specific phrasing that patients and physicians use.
The second component is a scoring rubric aligned to current REMS program documentation. Each AI output for a REMS-related query is scored against the specific requirements of the current program — not against the general clinical profile of the drug, and not against prior versions of the REMS. The scoring rubric should be updated every time the REMS is modified.
The third component is a cadence and escalation process. Weekly monitoring for complex REMS programs, monthly for simpler ones. Escalation criteria that distinguish between errors that are factually wrong about the drug’s clinical profile, errors that are wrong about current REMS requirements, and errors that are outdated because the REMS was modified after the model’s training cutoff.
The fourth component is a response capability. Not all AI misinformation about REMS programs can be corrected, but some of it can be reduced through the content and citation strategies described above. Companies need to know which AI errors they can address through published content and which they can only document and monitor.
The Broader Regulatory Question: Should AI Platforms Be Held to Pharmacovigilance Standards?
This is the policy question that is forming at the edge of AI governance and pharmaceutical regulation. When an AI platform gives a patient factually wrong information about how to use a drug, and that wrong information contributes to a clinical event, the regulatory question is: who bears responsibility? The drug manufacturer? The AI platform? The healthcare provider who recommended the AI tool? The patient who acted on it?
FDA Draft Guidance on AI in Drug Regulatory Decision-Making: What It Means for REMS
The FDA’s January 2025 draft guidance on AI use in regulatory decision-making focuses primarily on how sponsors can use AI in the drug development and approval process, not on how third-party AI platforms handle drug information. It establishes principles around AI validation, transparency, and human oversight that apply to sponsor activities. But those principles, applied to the REMS context, imply that any AI tool a sponsor uses to draft, review, or communicate REMS program materials must be validated for that purpose, with appropriate human oversight of outputs.
The EMA, which governs pharmaceutical regulation across the European Union, has not yet issued equivalent guidance specifically on AI and REMS-type programs, though its pharmacovigilance framework and the Annex 11 guidelines on computerized systems in GMP environments establish relevant precedents. The international dimension matters for companies with global REMS-like programs, where the regulatory requirements vary by jurisdiction and the AI models operate without geographic awareness.
What the Purolea Warning Letter Tells Pharma About AI and Regulatory Responsibility
The April 2026 Purolea warning letter established, in explicit regulatory text, that ‘AI does not excuse the failure to implement fundamental controls.’ That holding is likely to expand beyond manufacturing. The logic is straightforward: FDA-regulated activities require human oversight and expert judgment. Deploying AI to perform those activities without validating the AI’s outputs and maintaining human review does not transfer regulatory responsibility to the AI system. The company and its personnel bear the consequences.
For REMS-related communications, that logic implies that a sponsor cannot delegate to AI the accuracy of the safety information associated with its drug. Whether AI is used to draft patient materials, generate prescriber communications, monitor adverse event reports, or respond to medical information inquiries, the sponsor’s obligation to accuracy and regulatory compliance does not change. The AI is a tool. The obligation belongs to the company.
That is simultaneously a caution about careless AI adoption and an argument for careful AI deployment. A pharmaceutical company that builds structured, validated AI workflows for REMS monitoring and communication — with human expert review at each consequential step — can leverage AI’s speed and scale while maintaining the oversight that regulatory compliance requires. The companies that fail to build those workflows, and instead rely on general-purpose AI tools without governance, are exposing themselves to the same kind of enforcement exposure that Purolea encountered.
DrugChatter’s Role in AI Monitoring for REMS Drugs
DrugChatter tracks AI-generated drug information across the major LLM platforms, scoring outputs for factual accuracy, regulatory alignment, sentiment, and competitive framing. For drugs under active REMS programs, the platform’s query library can be configured to focus specifically on REMS-related questions — the prescriber queries, patient experience questions, and dispensing-related inquiries that are most likely to surface inaccurate REMS information.
The monitoring data that DrugChatter generates gives pharmaceutical brand and medical affairs teams a systematic view of what AI is telling the people who use their drugs. That view is not a replacement for the FDA’s REMS assessment reports, for the company’s pharmacovigilance program, or for the medical information team’s direct engagement with healthcare providers. It is the visibility layer that covers the gap between all of those programs and the AI channel where a growing share of drug information decisions are being made.
For a company with an active REMS program, that visibility layer is not a nice-to-have. It is a risk management function. The REMS was designed to ensure that the information environment around a high-risk drug is accurate, current, and operationally complete. AI has created a parallel information environment where none of those things are guaranteed. Monitoring that environment is how companies demonstrate that they take the REMS seriously in the channels where patients and physicians actually live.
Key Takeaways
- AI models — including ChatGPT, Gemini, Claude, and Perplexity — routinely misrepresent REMS requirements for high-risk drugs including isotretinoin, clozapine, mifepristone, opioid analgesics, and CAR T-cell therapies. The errors are not random; they are systematic, predictable, and tied to how AI training data weights regulatory documentation against patient forum content and health media coverage.
- The FDA’s April 2026 warning letter to Purolea Cosmetics Lab — the first to explicitly cite AI misuse in pharmaceutical manufacturing — established that AI does not transfer regulatory responsibility. Drug sponsors that use AI in any REMS-related function bear full accountability for the accuracy of AI-generated outputs.
- REMS programs change. The iPLEDGE program was modified in February 2026. The Clozapine REMS was removed in June 2025. The Opioid Analgesic REMS was modified in October 2024. AI models do not update on the same schedule as regulatory changes, creating periods where physicians and patients receive outdated safety information through AI channels.
- Different AI platforms describe the same REMS drug differently. Monitoring a single platform gives an incomplete picture. A systematic program covers ChatGPT, Gemini, Claude, Perplexity, and Copilot with platform-specific query batteries and scoring rubrics tied to current program documentation.
- AI REMS monitoring is a medical affairs and pharmacovigilance function, not a marketing one. The organizational home for this monitoring should be in the teams that own clinical accuracy accountability, with escalation paths to regulatory affairs and safety.
- Pharmaceutical companies can influence what AI says about their REMS programs by publishing clear, structured, current, machine-readable program documentation and earning high-authority citations. This is not about gaming AI — it is about ensuring that the most authoritative sources are the ones that shape AI outputs.
- The patient-facing risk is real. Patients asking AI chatbots about REMS drugs are receiving answers that range from incomplete to materially wrong. Companies that monitor these patient-query patterns can identify emerging compliance concerns before they appear in formal adverse event reporting.
Frequently Asked Questions
Can AI misrepresenting a REMS requirement create liability for a drug manufacturer?
No direct regulatory precedent as of mid-2026 holds a manufacturer liable for AI outputs it did not produce or distribute. But the FDA’s April 2026 Purolea warning letter establishes that companies cannot outsource regulatory compliance to AI systems. If a manufacturer uses AI to draft REMS-related communications that turn out to be inaccurate, the FDA’s current enforcement posture suggests the company — not the AI — bears responsibility for that inaccuracy. Manufacturers should treat REMS-related AI outputs, whether produced internally or observed in third-party platforms, as material that warrants active monitoring and, where the manufacturer has distributional control, expert human review.
Why do AI models get REMS requirements wrong even when the information is publicly available?
AI models learn from the full distribution of text available during training, not from authoritative sources alone. REMS documentation is precise and specific but relatively sparse on the public internet. Patient forum content, health news coverage, and secondary medical summaries are voluminous but imprecise. Models synthesize across all of these, with high-volume imprecise content often exerting more influence than low-volume precise content. Knowledge cutoffs compound the problem — REMS programs change after training data is collected, and models do not flag their own information as potentially outdated.
What is the difference between AI REMS errors for physicians versus patients?
Physician queries typically involve operational and prescribing specifics — enrollment steps, monitoring intervals, consequences of non-compliance. Patient queries typically involve the experience of being on a REMS drug — what to expect, what the requirements mean in daily life, what happens if something goes wrong. Both types carry clinical risk, but in different ways. A physician who misunderstands enrollment requirements may fail to complete required steps. A patient who misunderstands monitoring requirements may not comply with them. AI errors for both audiences are undermonitored and consequential.
How should pharmaceutical companies prioritize AI monitoring across their REMS drug portfolios?
Start with the programs that have changed most recently, because AI models are most likely to have outdated information about them. Then prioritize by ETASU complexity — programs with multi-step prescriber, pharmacy, and patient requirements carry higher error risk because there are more steps to get wrong. Drugs with significant patient-volume and high consumer AI query rates (isotretinoin, opioid analgesics, mifepristone) warrant more intensive monitoring than drugs with narrow specialist populations. Build a tiered monitoring cadence: weekly for complex, recently modified REMS programs; monthly for stable, lower-complexity programs.
Does Perplexity AI give more accurate REMS information than ChatGPT because it searches the web?
Perplexity’s web retrieval capability makes it more likely to surface current FDA documentation when a query triggers retrieval of official sources. Its outputs are also more verifiable — users can follow citations and check. But Perplexity’s accuracy advantage is query-dependent. Queries phrased in patient language (conversational, experience-focused) may not trigger official source retrieval. Queries about recently modified REMS programs may retrieve older documentation that is still indexed. Perplexity is more transparent about its sources than most AI platforms, which makes it easier to identify when the underlying source is outdated — but transparency is not the same as accuracy. All major platforms, including Perplexity, require active REMS-specific monitoring.






