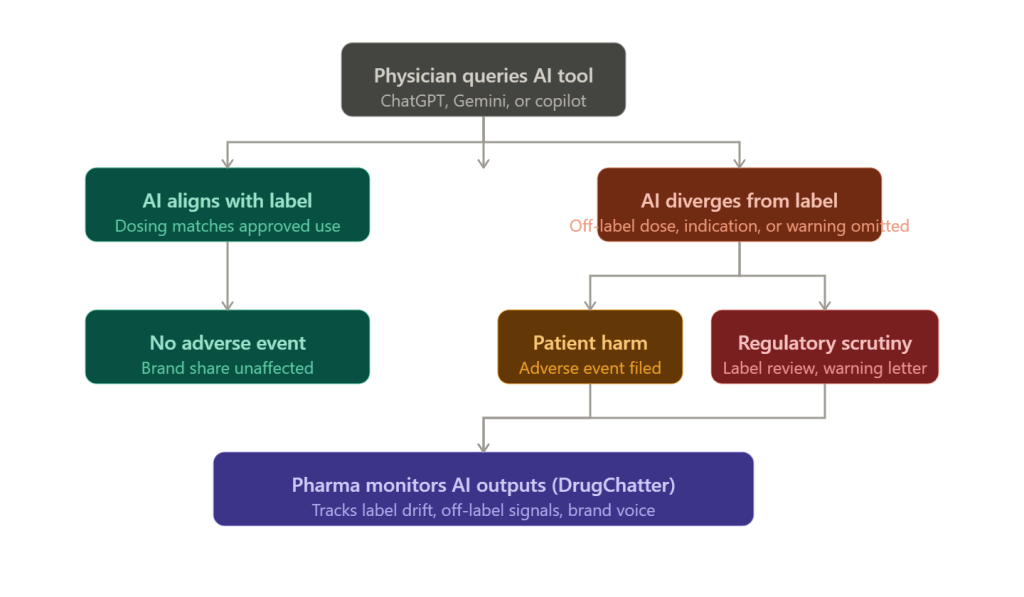
Physicians are increasingly asking AI chatbots how to dose drugs, manage contraindications, and choose between therapies. The AI answers. The FDA label sits unopened in a browser tab nobody clicked.
That gap, between what the label says and what the AI outputs, is becoming one of the most consequential compliance problems in the pharmaceutical industry. Drug companies built their regulatory submissions around a specific set of approved indications, dosing regimens, and safety warnings. Those submissions required years of clinical trials, millions of dollars, and a formal review process by the Food and Drug Administration. An AI system trained on PubMed abstracts, Reddit threads, UpToDate, and clinical forum posts can contradict all of it in 200 milliseconds.
This is not hypothetical. Multiple studies published between 2023 and 2025 found that large language models produce clinically significant errors when answering pharmacological questions, ranging from incorrect dosing to omitted black-box warnings. Physicians who trust those outputs and prescribe accordingly can trigger a chain of events, from adverse event filings to REMS violations, that lands squarely on the manufacturer’s doorstep even when the manufacturer did nothing wrong.
For pharma companies paying attention, this creates both a risk and an intelligence opportunity. Tools like DrugChatter now allow manufacturers to systematically track how AI systems describe their products, detect label drift in real time, and capture what prescribers actually hear when they query a model, which is increasingly different from what the approved label says.
The Prescribing Information Nobody Reads
Before 1970, drug labeling was chaotic. Package inserts were long, formatted inconsistently, and written for regulatory review rather than clinical use. The FDA began mandating structured prescribing information incrementally, culminating in the 2006 Physician Labeling Rule that introduced the Highlights section, a condensed summary of critical prescribing information that a physician could read in under two minutes.
The problem is they often don’t read it.
A 2022 survey published in the Journal of General Internal Medicine found that fewer than 15 percent of physicians reported regularly consulting the FDA-approved prescribing information before initiating a new drug therapy. The more common path runs through colleagues, continuing medical education, drug reps, and increasingly, digital tools.
AI tools are now part of that ecosystem. When Epic launched its AI assistant powered by GPT-4 in 2023, it was positioned as a clinical decision support tool. When Microsoft integrated Copilot into Teams channels used by hospital systems, clinical queries followed within weeks. When a physician in a busy urban emergency department types ‘what is the maximum dose of semaglutide for a 90 kg patient with CKD stage 3,’ the response they receive is not necessarily what Novo Nordisk’s label says.
The FDA approved Ozempic at doses up to 2 mg weekly. The prescribing information for patients with renal impairment includes specific pharmacokinetic monitoring guidance. Whether any given AI tool reproduces that guidance accurately, or omits it, or substitutes information from a European product monograph or a preprint paper, depends entirely on the model’s training data, system prompt, and retrieval architecture. Novo Nordisk has no control over any of those variables.
What AI Models Actually Say About Drugs
The research picture on AI pharmacology accuracy is not reassuring.
A study published in JAMA Network Open in late 2023 tested ChatGPT-3.5 and ChatGPT-4 on 100 standardized pharmacology questions drawn from board examination banks and clinical practice scenarios. GPT-4 achieved an accuracy rate of approximately 68 percent on dosing questions and 71 percent on drug interaction questions. On questions involving pregnancy category designations, it scored 54 percent, barely above chance.
A separate analysis published in Annals of Pharmacotherapy in early 2024 evaluated AI responses to questions about anticoagulation management, a domain where dosing errors carry severe bleeding risk. The authors found that Claude, GPT-4, and Gemini all produced at least one clinically dangerous recommendation when tested against 40 scenarios derived from real hospital cases. The errors were not random. They clustered around situations where label guidance conflicted with emerging clinical practice, which is precisely where an AI trained on recent literature would diverge from an approved label that had not been updated.
The structural reason for this divergence is not hard to identify. FDA-approved prescribing information is updated through a formal supplement submission process. A manufacturer who discovers a new drug interaction or wants to add a new dosing recommendation must file a Prior Approval Supplement or a Changes Being Effected supplement, depending on the significance of the change. That process takes months to years. Meanwhile, the clinical literature moves faster. By the time a warning appears on the label, hundreds of papers describing the underlying concern have already been indexed in PubMed, absorbed into AI training datasets, and synthesized into model responses.
This means AI models are often more current than labels in areas of active clinical research, and radically out of step with labels in areas where regulatory submissions have not kept pace with practice. Both directions carry risk. <blockquote> “AI tools in clinical settings are currently trained on an unvalidated mix of peer-reviewed literature, clinical forums, and patient advocacy content. None of that is FDA-approved prescribing information, and the distinction matters enormously when adverse event causality is being assessed.” <cite>— Drug Safety journal, 2024, citing analysis of AI pharmacology training corpora</cite> </blockquote>
The Regulatory Exposure Manufacturers Don’t See Coming
When a physician prescribes a drug off-label, the manufacturer is generally not liable for that decision as a matter of product liability law. The learned intermediary doctrine holds that a manufacturer’s duty to warn runs to the prescribing physician, not the patient directly, and that a physician who ignores or misapplies label guidance breaks the causal chain.
AI changes that analysis in ways courts have not yet fully resolved.
Consider a scenario that has already appeared in internal risk discussions at several large pharmaceutical companies: a physician uses an AI clinical decision support tool integrated into their EHR. The tool, queried about a drug, returns guidance that contradicts the approved label, perhaps omitting a contraindication or suggesting a dose that exceeds the approved maximum. The physician follows the AI guidance. A patient is harmed. Who bears liability?
The manufacturer faces at least three plausible exposure vectors. First, plaintiffs’ counsel could argue that the manufacturer’s own promotional materials, speaker programs, or sales force messaging contributed to an off-label impression that the AI then amplified and institutionalized. Second, if the manufacturer was aware that AI tools were systematically misrepresenting their product and failed to take corrective action, that awareness could be characterized as negligence. Third, some AI tools are built on licensed pharmaceutical data that manufacturers provided, creating a contractual and reputational entanglement.
The FDA has moved carefully in this space. The agency’s 2021 action plan on AI/ML-based software as a medical device (SaMD) and its 2023 draft guidance on AI-enabled devices both acknowledge the challenge of ensuring that AI outputs in clinical settings conform to approved labeling. But neither document creates a clear enforcement mechanism targeting manufacturers whose products are being misrepresented by third-party AI systems.
That regulatory ambiguity is itself a risk factor. When the rules are unclear, enforcement tends to be reactive, and reactive enforcement typically follows a high-profile adverse event. Manufacturers who are not monitoring how AI systems describe their drugs will be the ones caught unprepared when that event occurs.
GLP-1 Drugs and the AI Description Problem
No drug class illustrates the AI prescribing gap more vividly than the GLP-1 receptor agonists.
Semaglutide (Ozempic, Wegovy), liraglutide (Victoza, Saxenda), and tirzepatide (Mounjaro, Zepbound) are among the most-queried drugs in AI clinical tools as of 2024 and 2025. The off-label use of diabetes-approved formulations for weight loss was already widespread before Wegovy received its FDA approval in June 2021. By 2023, that off-label use had created a shortage that forced the FDA to place both Ozempic and Wegovy on its drug shortage list.
Throughout this period, AI tools were actively contributing to prescriber confusion about which formulation to use, what doses were appropriate, and whether the diabetes-approved and obesity-approved formulations were interchangeable. They are not interchangeable in terms of regulatory status, but the pharmacological similarity is real, and AI models trained on clinical literature rather than regulatory documents consistently fail to maintain the distinction.
Novo Nordisk’s approved label for Ozempic specifies doses of 0.5 mg, 1 mg, and 2 mg weekly for type 2 diabetes management. The approved label for Wegovy specifies a different titration schedule reaching 2.4 mg weekly for chronic weight management. A physician who asks an AI tool ‘can I use Ozempic for weight loss at 2.4 mg’ is likely to receive an answer that reflects clinical practice rather than approved labeling, because clinical practice had moved ahead of regulatory approval.
Novo Nordisk’s pharmacovigilance teams have had to work in an environment where AI-driven prescribing behavior is creating adverse event patterns that do not map cleanly onto the approved indication, making causality assessment and label update decisions significantly more complicated.
Eli Lilly faces similar dynamics with tirzepatide. Mounjaro (tirzepatide for diabetes) and Zepbound (tirzepatide for obesity) were approved separately, with the obesity indication arriving in November 2023. AI tools, queried about tirzepatide dosing for weight loss before and after that approval, produced answers that reflected an evolving and sometimes internally inconsistent picture. Lilly’s medical affairs function had no automated way to track what prescribers were hearing from those tools.
Oncology: Where Label Accuracy Is a Life-or-Death Matter
If the GLP-1 situation is commercially damaging, the oncology equivalent is clinically dangerous.
Cancer drugs carry narrow therapeutic windows, complex dosing adjustments based on renal function and hepatic metabolism, and black-box warnings that exist because the underlying risks are severe. The approved labels for drugs like pembrolizumab (Keytruda), osimertinib (Tagrisso), and venetoclax (Venclexta) are among the most detailed and carefully worded documents in pharmaceutical regulation.
Pembrolizumab’s label runs to more than 50 pages. It specifies immune-mediated adverse reactions, infusion-related reactions, and a specific set of contraindications in solid organ transplant patients, an area where clinical practice continues to evolve and where AI models trained on oncology literature may suggest use where the label is silent or cautious.
A 2024 analysis in the Journal of Clinical Oncology evaluated AI responses to 60 oncology prescribing scenarios. The authors found that all four tested AI systems, including GPT-4 and Gemini 1.5, produced at least some responses that recommended treatment combinations not supported by the current FDA-approved label, often citing published clinical trial data that had not yet been incorporated into the label through a regulatory supplement.
This is not the AI systems being reckless. It reflects the fundamental tension between evidence-based medicine, which moves at the speed of publication, and regulatory medicine, which moves at the speed of formal submission. But the manufacturer of pembrolizumab, Merck, bears reputational and potentially regulatory consequences when prescribers attribute their dosing decisions to what ‘the AI said about Keytruda,’ regardless of whether Merck had any involvement with that AI system.
REMS Programs and the AI Awareness Gap
Risk Evaluation and Mitigation Strategies represent the FDA’s most aggressive intervention in drug distribution and prescribing. REMS programs exist for drugs whose approved use is so tightly constrained by safety considerations that standard labeling is insufficient to prevent harm.
Clozapine, the antipsychotic used in treatment-resistant schizophrenia, requires enrollment in the Clozapine REMS program before a single prescription can be dispensed. The program mandates absolute neutrophil count monitoring at defined intervals, with prescribing and dispensing locked out if the monitoring is not current. The REMS system is the enforcement mechanism that makes those monitoring requirements real.
Isotretinoin (Absorica, Claravis, and others) operates under the iPLEDGE REMS, which requires pregnancy testing, contraceptive counseling, and a monthly attestation process for patients of childbearing potential. The program was designed after thalidomide-era teratogenicity concerns prompted Congress to require manufacturers to demonstrate that risk management was in place before certain drugs could remain on the market.
When a physician or patient asks an AI tool about clozapine or isotretinoin, the response they receive may accurately describe the pharmacology, the approved indications, and the common adverse effects without ever mentioning the REMS requirement. This is not because the AI is wrong about the pharmacology. It is because REMS information is procedural, regulatory, and not consistently represented in the clinical literature on which AI models are trained.
A pharmacist who catches a clozapine prescription from a prescriber unfamiliar with the REMS may prevent a dispensing error. An AI tool that fails to surface the REMS requirement in its initial response may create a situation where neither the prescriber nor the patient is prepared for the enrollment process, leading to delays, frustration, and in some cases prescriber abandonment of a drug that was clinically appropriate.
For manufacturers operating drugs under REMS, the AI awareness gap is a direct program integrity issue. The FDA expects REMS programs to be effective, and effectiveness is measured partly by whether prescribers understand the requirements. If AI tools are creating an information environment where REMS requirements are invisible, manufacturers may face pressure to expand their REMS communications in ways they had not anticipated.
Brand Voice in the Age of AI-Mediated Prescribing
The traditional pharmaceutical brand exists in a carefully managed information ecosystem. Medical affairs controls the published science through publications planning and symposia. Commercial controls the messaging through promotional review committees and the FDA-regulated promotional materials approval process. Sales forces deliver specific, approved messages through detail calls governed by company SOPs and federal regulations.
AI tools have created a parallel information channel that operates entirely outside this system.
When a physician asks ChatGPT to compare apixaban (Eliquis) and rivaroxaban (Xarelto) for a patient with atrial fibrillation and moderate renal impairment, the AI synthesizes an answer from its training data. That answer may accurately reflect the clinical literature, may over-represent one drug or the other based on the volume of studies in the training data, may miss label-specific dosing adjustments for renal function, or may reflect promotional language that appeared in trade publications or patient advocacy materials without the physician realizing it.
Bristol Myers Squibb and Pfizer, who co-commercialize Eliquis, have no visibility into how AI tools describe their drug relative to competitors unless they are actively monitoring those descriptions. Bayer and Johnson & Johnson, who co-commercialize Xarelto, are in the same position.
Brand share of voice has historically been measurable through sales force call metrics, prescription data, and market research. The AI channel is harder to measure because it is decentralized, interaction-level data is not available to manufacturers, and the volume of physician-AI interactions is growing faster than any monitoring system built on traditional methods can track.
This is where specialized tools like DrugChatter enter the picture. By systematically querying AI systems with standardized prompts about specific drugs, formulations, and therapeutic comparisons, DrugChatter generates data on how those drugs are described, what competitive framing appears, what safety information is included or excluded, and whether AI responses are consistent with approved prescribing information. That data is actionable: it identifies where label drift is occurring, which AI systems are most problematic for a given drug, and what corrective communication may be needed.
The Pharmacovigilance Dimension
Adverse event detection is already under strain. The FDA’s MedWatch system captures a fraction of actual adverse events, with post-marketing surveillance studies typically estimating underreporting rates between 90 and 99 percent. Adding AI-mediated prescribing as a causality variable makes the problem more complex.
When a patient experiences an adverse event after taking a drug that was prescribed based on AI guidance that differed from the approved label, the adverse event report typically does not capture the prescribing decision pathway. The MedWatch form asks about the drug, the dose, the indication, and the adverse event. It does not ask whether the prescriber consulted an AI tool.
This creates a systematic gap in pharmacovigilance data. Manufacturers building their signal detection models on adverse event data will not see the AI-mediated prescribing pathway unless they specifically look for it. And looking for it requires knowing that AI tools are describing their drugs in ways that could affect prescribing decisions, which requires the kind of active monitoring that most pharmacovigilance operations are not yet performing.
The FDA has begun acknowledging this gap. The agency’s 2024 discussion paper on AI in drug development mentions the challenge of pharmacovigilance in an environment where AI tools are influencing prescribing, but stops short of specific guidance. Manufacturers who get ahead of this issue by building monitoring capabilities now will be better positioned to provide meaningful data if the FDA moves toward requiring it.
What Proactive Manufacturers Are Actually Doing
A small number of large pharmaceutical companies have begun treating AI monitoring as a medical affairs function rather than an IT problem.
The practical approach involves three components. First, systematic querying of major AI platforms (ChatGPT, Gemini, Claude, Copilot, and specialty clinical tools like Doximity’s AI and Epic’s AI assistant) with standardized prompts designed to elicit drug descriptions, dosing guidance, and competitive comparisons. Second, comparison of AI outputs against approved prescribing information, with discrepancies categorized by severity: omitted black-box warnings, incorrect dosing, wrong indication, misleading competitive framing. Third, integration of those findings into medical affairs strategy, informing where corrective scientific communications are needed and where label update supplements should be prioritized.
Several companies have also begun engaging directly with AI platform developers, providing approved prescribing information in machine-readable formats and requesting that specific labeling elements, particularly REMS requirements and black-box warnings, be surfaced in responses to relevant clinical queries. This engagement is voluntary and inconsistent, but it reflects recognition that passive approaches are insufficient.
DrugChatter has built infrastructure around the systematic monitoring component, allowing pharmaceutical companies to track AI outputs across multiple platforms at scale, with alerts when significant label drift is detected. For a drug with hundreds of possible query variations and multiple AI platforms to monitor, manual monitoring is not feasible. Automated monitoring with human review of flagged outputs is the only approach that scales.
The return on investment for this capability is difficult to quantify precisely because the harm being prevented is counterfactual. But the calculus becomes clearer when you consider the cost components of the alternative: a single serious adverse event that can be traced to AI-mediated prescribing can generate litigation costs exceeding $10 million, FDA correspondence that requires C-suite attention, and reputational damage to a brand that took years to build.
The Learned Intermediary Doctrine Under Pressure
Product liability attorneys specializing in pharmaceutical cases have begun documenting the AI prescribing pathway as a potential theory of manufacturer liability, and their reasoning deserves attention from legal and regulatory affairs teams.
The learned intermediary doctrine was developed in a world where the physician was the sole filter between the manufacturer’s warning and the patient’s decision to take a drug. The physician read the label (or was supposed to), evaluated the patient’s specific circumstances, and made a prescribing decision. If the label was adequate and the physician ignored it, the manufacturer was not liable for the physician’s error.
AI tools create a new intermediary, one that the manufacturer did not train, does not control, and cannot discipline. When a physician relies on an AI tool’s response instead of reading the label, the traditional model breaks down. Plaintiffs’ counsel can argue that the manufacturer knew AI tools were being used to guide prescribing, knew those tools were misrepresenting the drug’s safety profile, and failed to take reasonable steps to correct the misrepresentation.
That argument has not yet been tested in a published decision. But it does not need to be tested to be expensive. Discovery in pharmaceutical litigation routinely requires manufacturers to produce internal communications about drug risks and safety signals. If those communications show that the company was aware of AI misrepresentation of its product and made no effort to correct it, that awareness becomes a liability exhibit.
The mirror image is also true. If a manufacturer can demonstrate that it had active monitoring in place, that it identified and documented AI-related label drift, and that it took specific corrective actions including engaging with platform developers and updating its medical communications strategy, that documentation becomes a defense exhibit. The monitoring capability is not just a risk management tool. It is evidence of reasonable manufacturer conduct in a new regulatory environment.
FDA’s Evolving Position and What It Signals
The FDA has been characteristically deliberate in its approach to AI and prescribing. The agency moves through guidance documents, discussion papers, and public workshops rather than through enforcement letters and consent decrees, at least initially.
The trajectory of FDA signaling on AI is becoming clearer. The 2021 AI/ML action plan, the 2023 draft guidance on AI-enabled devices, and a series of workshop proceedings from 2022 through 2024 collectively establish a regulatory posture that recognizes AI as a medical device when it meets the statutory definition of a device under the Food, Drug, and Cosmetic Act, but that struggles with the question of how to regulate AI tools that provide clinical information without making specific diagnostic or therapeutic recommendations.
A chatbot that says ‘the FDA-approved dose of methotrexate for rheumatoid arthritis is 7.5 to 20 mg weekly’ is providing clinical information but probably not making a clinical decision. A chatbot that says ‘for your patient with these specific characteristics, I recommend starting methotrexate at 10 mg weekly’ is making a recommendation that looks more like a clinical decision. The boundary between those two outputs is what the FDA is trying to define, and where that boundary lands will determine how much regulatory pressure flows toward AI developers versus how much flows toward pharmaceutical manufacturers who fail to ensure their label information is accurately represented.
Manufacturers who treat AI monitoring as a compliance function today, rather than waiting for FDA guidance to make it mandatory, will have built both the capability and the regulatory goodwill that accompanies proactive action.
Signal Detection: The New Medical Affairs Priority
Medical affairs organizations have historically focused on scientific exchange, publication strategy, and medical education. The AI prescribing environment is creating a new function within medical affairs: active signal detection for label drift in AI systems.
Signal detection in this context means identifying, at scale and in real time, the points where AI descriptions of a drug diverge from approved prescribing information. Those divergence points are not randomly distributed. They cluster around specific question types: dosing in special populations, drug-drug interactions, use in pregnancy and lactation, competitive comparisons, and emerging data not yet incorporated into the label.
Each divergence point represents a potential prescribing error, a potential adverse event, and a potential regulatory inquiry. Medical affairs organizations that build detection capabilities around these divergence points will be able to prioritize their corrective communications more precisely, focusing resources on the AI responses that are generating the most clinical risk rather than attempting to address all AI activity simultaneously.
The intelligence value extends beyond risk management. When AI tools systematically describe a competitor’s drug in terms that are more favorable than the approved label warrants, that represents a competitive intelligence signal. When AI tools describe a company’s own drug in terms that undersell the clinical benefit relative to the label, that represents a messaging gap that medical affairs can address. The monitoring capability that protects against regulatory risk also generates insights that inform commercial strategy.
Key Takeaways
AI tools are now a primary prescribing information source for a significant proportion of physicians, and the information those tools provide routinely diverges from FDA-approved labeling in clinically significant ways.
Manufacturers bear indirect but real risk from that divergence, including product liability exposure, REMS program integrity concerns, and pharmacovigilance data gaps that complicate adverse event causality assessment.
The learned intermediary doctrine, the traditional manufacturer liability shield, is under doctrinal pressure in cases where AI tools mediate between the label and the prescribing decision.
Active monitoring of AI drug descriptions, through tools like DrugChatter and systematic medical affairs protocols, is the most actionable response available to manufacturers today, both as a risk management measure and as a source of competitive intelligence.
The FDA’s regulatory posture on AI and prescribing is still forming, which means manufacturers who build monitoring capabilities and engage proactively with the issue will have the best opportunity to shape the regulatory expectations that eventually emerge.
Brand share of voice is increasingly determined not just by what sales forces and promotional materials say, but by what AI tools output when physicians query them, and manufacturers who are not tracking that channel are operating with a significant blind spot.
FAQ
Q: Can a pharmaceutical manufacturer be held liable for an adverse event that resulted from a physician following AI guidance instead of the approved label?
This is the central unresolved question in pharmaceutical AI liability. The traditional learned intermediary framework would typically shield the manufacturer if the label was adequate and the physician chose to rely on a different information source. But courts have not yet addressed situations where a manufacturer was demonstrably aware that AI systems were systematically misrepresenting its product and failed to act. That awareness-plus-inaction scenario is where liability arguments are most likely to gain traction. Manufacturers who actively monitor and document AI outputs, and who take corrective action when significant divergence is identified, are in a substantially stronger defensive position.
Q: Why do AI models get drug information wrong if they have access to FDA label databases?
Access to FDA label text and training on FDA label text are different things. Most large language models were not specifically trained to weight FDA prescribing information more heavily than clinical literature. A model trained on PubMed, clinical guidelines, textbooks, and clinical forum data will produce responses that reflect the aggregate of that literature, which often differs from the approved label in meaningful ways. Even retrieval-augmented systems that can access current label databases may blend label information with other sources in ways that dilute or contradict specific label provisions. The problem is architectural, not a simple data gap.
Q: How does DrugChatter differ from standard pharmacovigilance signal detection?
Traditional pharmacovigilance signal detection focuses on adverse event reports, medical literature, and social media patient reports to identify new safety signals emerging in the post-market environment. DrugChatter is oriented in the opposite direction: it monitors how AI systems describe drugs prospectively, before adverse events occur, to identify the information gaps and errors that might contribute to future prescribing decisions. The two functions are complementary. Pharmacovigilance detects harm after it has occurred; AI monitoring identifies the information environment that shapes prescribing decisions before harm occurs.
Q: What REMS program categories are most exposed to AI awareness gaps?
REMS programs with complex enrollment, monitoring, and certification requirements are most exposed, because those procedural requirements are least likely to appear in the clinical literature that trains AI models. The clozapine REMS (absolute neutrophil count monitoring and pharmacy certification), the isotretinoin iPLEDGE program (monthly pregnancy testing and contraceptive attestation), the TIRF REMS Access program for transmucosal immediate-release fentanyl products, and the opioid REMS programs for extended-release formulations all involve multi-step prescriber and patient enrollment that AI tools routinely fail to describe adequately. Manufacturers operating drugs under these programs should treat AI monitoring as a REMS communications audit function.
Q: Is there regulatory precedent for the FDA requiring manufacturers to correct AI misinformation about their products?
No direct precedent exists as of 2025. The FDA has statutory authority under Section 301 of the FDCA to prohibit false or misleading labeling, but that authority runs to labeling that the manufacturer controls or disseminates. Third-party AI outputs are not manufacturer labeling in the traditional sense. However, if a manufacturer provides drug information to an AI platform in a licensed data relationship, and that platform uses the information in ways the manufacturer knows to be inaccurate, FDA could theoretically characterize the manufacturer’s continued participation in that relationship as contributing to misbranding. That theory has not been tested, but it illustrates the importance of documenting manufacturer efforts to ensure accuracy in AI systems where they have any contractual relationship.