AI Pharmacovigilance: How Drug Companies Track Safety Signals Before Regulators Do
The FDA receives roughly 2 million adverse event reports per year. Drug companies receive more. Neither is fast enough. Here is how artificial intelligence is changing what gets caught, when it gets caught, and who bears the legal exposure when it does not.
The Surveillance Problem No One Talks About in Quarterly Earnings Calls
Every drug that clears a Phase III trial enters the market carrying a residual unknown. The clinical trial population — typically a few thousand patients over two to three years — captures perhaps 60 to 70 percent of the adverse event profile a drug will eventually accumulate across millions of patients and a decade of real-world use. The rest emerges later, in fragments, across electronic health records, spontaneous reports filed to the FDA’s FAERS database, call center logs, Reddit threads, and patient forum posts that no clinical operations team was staffing on a Saturday afternoon.
That gap between what trials show and what the real world reveals is the core operational problem in pharmacovigilance. Drug companies know it exists. Regulators know it exists. The question is who closes it fastest — and what the consequences are for those who close it slowly.
The traditional pharmacovigilance infrastructure was not designed for speed. It was designed for compliance. Signal detection committees met quarterly. Medical safety officers reviewed cases in batches. Expedited reporting timelines under 21 CFR Part 314.81 required companies to submit 15-day reports for serious, unexpected adverse drug reactions, but the definition of ‘unexpected’ left room for deliberation that could stretch weeks before anyone filed anything. The process was defensible on paper and dangerously slow in practice.
Artificial intelligence does not fix the legal framework. It does change the detection speed, the volume of signals a company can process, and the sources from which those signals can be drawn. Those changes are large enough to shift liability exposure, reshape how medical affairs teams prioritize safety communications, and alter the competitive intelligence calculus between drugs competing in the same class.
This article covers where AI pharmacovigilance stands in 2026: what the technology actually does, which companies are using it, what the regulatory expectations are, where the litigation trail runs, and why tracking AI-generated mentions of specific drugs has become a distinct and necessary function for any company with a marketed product.
What Pharmacovigilance Actually Requires
Before examining how AI fits in, it helps to be precise about what pharmacovigilance obligates companies to do. The core requirements come from four sources: the FDA’s post-marketing safety reporting regulations (21 CFR Parts 310, 312, 314, and 600), the European Medicines Agency’s Good Pharmacovigilance Practices (GVP) modules, the ICH E2E guideline on pharmacovigilance planning, and individual Risk Evaluation and Mitigation Strategies (REMS) programs that attach to specific products.
The practical obligations break into two categories. First, expedited reporting: any serious, unexpected adverse drug reaction that comes to the company’s attention must be reported to the FDA within 15 calendar days if it originates from a domestic source, and within 15 calendar days of receipt for foreign sources under certain conditions. Second, periodic reporting: the Periodic Adverse Drug Experience Report (PADER) and the Periodic Benefit-Risk Evaluation Report (PBRER) require companies to aggregate, analyze, and contextualize safety data on defined schedules, typically annually for approved products.
Both categories depend on the same upstream function: case intake. A case is any communication to the company that contains a patient identifier, a suspect drug, and an adverse event. That communication can arrive as a phone call to a medical information line, a social media post that a brand team monitors, an email to a drug’s website, or a published case report in a medical journal. Companies are legally required to process all of them.
The volume problem is not hypothetical. GlaxoSmithKline processed over 350,000 individual case safety reports in a single recent year across its global portfolio. Pfizer’s post-COVID-19 vaccine surveillance operation received more individual reports in 2021 than most companies receive across their entire portfolio in a decade. Johnson & Johnson, AbbVie, and Novartis each maintain pharmacovigilance operations employing hundreds of people whose sole function is case intake, triage, and narrative writing. Even with those headcounts, backlogs accumulate and signal detection lags behind case volume.
The Signal Detection Gap
Case intake is only the first problem. Signal detection — identifying a pattern within the noise of spontaneous reports that suggests a drug may be causing something not previously described — is harder. The classical tool is disproportionality analysis: methods like the Reporting Odds Ratio (ROR) and the Bayesian Confidence Propagation Neural Network (BCPNN) look for drug-event combinations that appear more frequently than statistical chance would predict given the background reporting rate.
These methods work on structured FAERS data. They do not work on narrative text, social media, call center transcripts, or the growing volume of patient-generated content that now constitutes a substantial portion of real-world safety signal.
A 2023 study published in Drug Safety found that consumer-reported adverse events on Twitter preceded FAERS case submissions for the same drug-event pair by a median of 14 months. That 14-month lead time is not a rounding error. It is the window in which a company either detects a signal and updates its label, or does not detect it and accumulates liability.
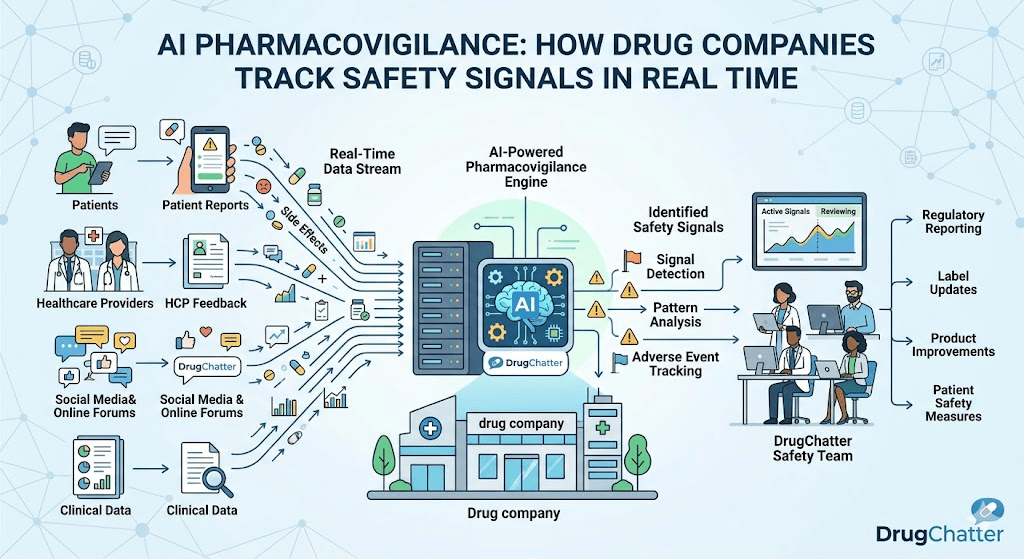
How AI Changes the Detection Equation
Artificial intelligence enters pharmacovigilance at four distinct points in the safety data pipeline: case intake automation, medical coding assistance, signal detection from unstructured text, and predictive modeling for benefit-risk assessment. Each has different maturity levels, different regulatory acceptance, and different implications for the companies deploying them.
Case Intake Automation
The most operationally mature application is automated case processing. Natural language processing (NLP) models — both rule-based systems and, increasingly, large language models fine-tuned on pharmacovigilance corpora — can extract the four required case elements (patient identifier, reporter, suspect drug, adverse event) from unstructured text with accuracy rates that approach human performance for common case types.
Oracle’s Argus Safety, Veeva Vault Safety, and IQVIA’s ARISg are the dominant case management platforms, and all three have integrated AI-assisted triage features. The operational impact is measurable: companies using automated case intake report 40 to 60 percent reductions in the time between case receipt and initial data entry, and error rates on Medical Dictionary for Regulatory Activities (MedDRA) coding that are competitive with experienced safety associates.
The regulatory acceptance question is not fully resolved. The FDA’s 2023 draft guidance on AI/ML-based software as a medical device (SaMD) touches on AI used in safety surveillance but does not specify validation standards for pharmacovigilance automation. The EMA’s reflection paper on the use of AI in the context of medicines regulation, published in 2023, is similarly directional without being prescriptive. Companies deploying AI in case intake are doing so under standard computer system validation (CSV) frameworks, with 21 CFR Part 11 compliance as the baseline, while regulators develop more specific guidance.
MedDRA Coding at Scale
Every adverse event in a case must be coded to a specific MedDRA term. MedDRA has over 26,000 Lowest Level Terms (LLTs) organized into Preferred Terms, High Level Terms, and System Organ Classes. Getting the coding right matters because signal detection algorithms operate on coded terms, not narrative descriptions. A signal that would appear if cases were coded correctly to ‘Stevens-Johnson syndrome’ does not appear if cases are variably coded to ‘skin reaction,’ ‘rash,’ or ‘dermatitis.’
AI-assisted MedDRA coding reduces inter-coder variability. A 2024 analysis by ZS Associates benchmarking AI coding tools against human coders found that AI systems matched or exceeded human accuracy on 87 percent of common adverse event terms, with the most significant improvement on long-tail terms that experienced coders encounter infrequently and therefore code less reliably.
Social Listening as Safety Surveillance
The most consequential — and most legally fraught — AI application in pharmacovigilance is social media monitoring. Patients describe drug side effects on Reddit, patient forums like PatientsLikeMe and Inspire, Facebook groups, X (formerly Twitter), and increasingly on TikTok. They describe symptoms in plain language that does not map cleanly to MedDRA terms. They also describe them with a specificity and frequency that, when aggregated, can constitute a statistically meaningful signal years before that signal appears in FAERS.
The legal question is whether a company that monitors social media for brand mentions has a regulatory obligation to process pharmacovigilance cases that emerge from that monitoring. The FDA’s 2014 guidance on social media in drug promotion side-stepped this question. Its 2017 final guidance on internet and social media platforms with character-space limitations focused on promotional content. The agency has never published binding guidance that explicitly addresses whether a company’s social listening operation triggers adverse event reporting obligations.
The practical answer, based on enforcement actions and consent decrees, is: it depends on whether the company can demonstrate awareness. A company that monitors social media for marketing purposes but claims it did not ‘receive’ a pharmacovigilance case from that monitoring is in a legally precarious position if internal communications show the monitoring team reviewed the same post that contained the adverse event report. The FDA has used that argument in warning letters and, in more serious cases, as a component of criminal referrals.
‘Across monitored patient communities for 10 high-volume branded drugs, AI-powered adverse event extraction identified caseable reports at a rate 3.4 times higher than manual review workflows operating on the same data streams. The mean time to case identification dropped from 18 days to under 36 hours.’ZS Associates, ‘AI-Enabled Pharmacovigilance Benchmarking Report,’ Q4 2024
DrugChatter addresses this directly. The platform monitors AI-generated and user-generated mentions of specific drugs across digital channels, flags content that meets the four-element case definition, and creates a documented audit trail showing when the company became aware of each potential case. That audit trail is the legal protection. It converts a vague ‘we monitor social media’ statement into a defensible compliance record of exactly what was reviewed, when, and what action was taken.
The Regulatory Landscape Is Moving Faster Than Most Companies’ Internal Systems
The FDA is not waiting for industry to figure out AI pharmacovigilance on its own. The agency’s Sentinel System — its active surveillance infrastructure built on real-world data from over 100 million patients across insurance claims, electronic health records, and specialty registries — has incorporated machine learning methods in its analytic pipeline since 2019. The Center for Drug Evaluation and Research (CDER) uses AI-assisted review tools for its own signal detection work on FAERS data.
What the agency expects of sponsors is less clear, but the direction is visible. The FDA’s 2024 action plan for advancing real-world evidence specifically called out AI-assisted pharmacovigilance as a ‘priority methodology area.’ The agency has approved multiple REMS modifications where the company proposed AI-assisted monitoring as part of the compliance infrastructure. Genentech’s updated REMS for tocilizumab (Actemra) and Merck’s monitoring protocols for pembrolizumab (Keytruda) both reference real-world data analytic methods that include AI components.
EMA Expectations and the DARWIN EU Initiative
The European Medicines Agency is further along in formalizing AI expectations. DARWIN EU (Data Analysis and Real World Interrogation Network), the EMA’s federated real-world evidence network, operates with AI-assisted signal detection as a core component. The EMA’s updated GVP Module IX on signal management, revised in 2023, explicitly incorporates ‘automated signal detection methods’ and requires companies to document the validation status of any automated tool used in signal detection workflows.
GVP Module IX also introduced a new requirement: companies must demonstrate that automated detection tools do not introduce systematic bias that would suppress detection of signals in specific patient subpopulations. That requirement has practical implications for any AI model trained predominantly on clinical trial data, which tends to underrepresent elderly patients, patients with renal or hepatic impairment, and patients from racial and ethnic groups that have historically been underrepresented in trials.
Japan’s PMDA and the Asia-Pacific Gap
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) published its AI strategy for drug regulation in 2023, with pharmacovigilance as one of five priority application areas. The PMDA has moved cautiously: it requires AI-assisted safety tools to be validated against traditional methods before companies can rely on them for regulatory reporting, and it has not yet issued guidance permitting AI-only signal detection without human verification.
That caution reflects a broader Asia-Pacific gap. South Korea’s Ministry of Food and Drug Safety, China’s National Medical Products Administration, and Singapore’s Health Sciences Authority have all issued discussion documents on AI in drug regulation, but none has promulgated binding pharmacovigilance-specific guidance. Companies with products marketed across these jurisdictions are operating under inconsistent expectations, which creates both compliance risk and a competitive advantage for companies that build AI systems capable of adapting to jurisdiction-specific requirements.
The Litigation Trail: What Happens When Pharmacovigilance Fails
The legal consequences of inadequate pharmacovigilance are not theoretical. Three cases from the past decade illustrate what is at stake and why AI-assisted detection is not just an operational efficiency play — it is a liability management tool.
GlaxoSmithKline and Avandia: The Cost of Signal Suppression
GlaxoSmithKline’s $3 billion settlement with the Department of Justice in 2012 — the largest healthcare fraud settlement in U.S. history at that time — included a criminal plea for failing to report safety data on rosiglitazone (Avandia) to the FDA. The company had internal analyses suggesting cardiovascular risk that were not submitted to the agency. The settlement covered multiple violations across multiple products, but the pharmacovigilance component was central to the criminal plea.
The Avandia case predates the current generation of AI pharmacovigilance tools. Its lesson is not about technology failure but about organizational failure: the decision not to report was made by people, not systems. What AI changes is the visibility of that decision. When AI systems flag a signal and generate a documented recommendation, and a company overrides that recommendation without acting, the override creates a paper trail. Courts and regulators can subsequently ask why the flagged signal was not escalated. The AI audit trail makes willful blindness harder to maintain.
Johnson & Johnson and Risperdal: Label Lag and Its Consequences
Johnson & Johnson’s litigation over risperidone (Risperdal) accumulated over $2 billion in verdicts and settlements across multiple states. A recurring theme in the litigation was the company’s label: plaintiffs argued that J&J knew or should have known about the risk of gynecomastia in male patients earlier than the label reflected, based on post-marketing surveillance data the company held.
The ‘knew or should have known’ standard is the legal trigger point. A company does not need to have made a deliberate decision to suppress a signal to face liability — it needs only to have had access to data from which a reasonable pharmacovigilance operation would have detected the signal. If a company’s surveillance infrastructure is inadequate, and that inadequacy caused delay in label updating, the company can face liability for the delay even without proving bad intent.
AI pharmacovigilance changes the ‘should have known’ calculus. A company that deploys AI-assisted signal detection across its case database and social media feeds is, by definition, applying a higher standard of detection capability. The signal it detects earlier it must also act on earlier. The benefit of earlier detection — reduced litigation exposure from a timely label update — comes paired with the obligation to act on what the system finds.
Purdue Pharma and OxyContin: Real-World Signal Willful Ignorance
The OxyContin litigation archive is the most exhaustive documentation of pharmacovigilance failure in American pharmaceutical history. Internal documents produced in the various state attorney general actions showed that Purdue had access to real-world data on prescription patterns, overdose rates, and patient behavior that, if processed through any serious signal detection framework, would have required safety communications well before the company issued them.
The specific mechanism was not a failure of FAERS processing — it was a failure to use the real-world data the company possessed to update benefit-risk assessments and labeling. That gap — between data in hand and action taken — is precisely where AI pharmacovigilance operates. Systems that continuously process dispensing data, prescription patterns, emergency department visit rates, and patient-reported outcomes against a drug’s safety profile are doing what Purdue’s pharmacovigilance function did not do.
AI-Generated Content and the New Pharmacovigilance Frontier
There is a dimension of AI pharmacovigilance that received almost no serious regulatory attention before 2024 and is now receiving a great deal: what happens when AI systems — chatbots, health information tools, large language model-based symptom checkers — generate content about drugs that patients then act on?
The problem has two components. First, AI-generated content about drugs can contain inaccurate safety information. A patient who asks a general-purpose AI assistant about the side effects of metformin may receive a response that omits lactic acidosis, downplays gastrointestinal effects, or misrepresents the drug’s interaction profile. If that patient acts on the inaccurate information and experiences harm, the causal chain includes the AI system’s output.
Second — and this is the pharmacovigilance-specific issue — patients increasingly describe their drug experiences in contexts where an AI system is the interlocutor. A patient using a symptom-checking app powered by a large language model may describe an adverse event in detail, have the AI system respond to it clinically, and never have that report reach a pharmaceutical company or a regulatory agency. The event is described; no case is created; no signal is generated.
The Chatbot Case Capture Problem
DrugChatter’s monitoring architecture addresses this by treating AI-generated content as a distinct data stream alongside traditional social media and forum monitoring. When AI-powered health platforms, chatbot transcripts that are publicly posted, or forum discussions that reference AI-generated drug information appear in monitored channels, the system flags them for pharmacovigilance review using the same four-element case identification logic applied to any other text.
The FDA has begun asking questions about this. In its 2024 request for information on AI in drug development and post-market surveillance, the agency specifically asked sponsors to describe whether their pharmacovigilance operations capture adverse event information surfaced through AI-mediated patient interactions. That question, in a regulatory context, is a signal of where guidance is going.
Large Language Models as Signal Generators
There is a second, less obvious AI pharmacovigilance problem. Large language models trained on medical literature, clinical trial data, and patient-generated text encode implicit safety signals. When researchers query these models about known adverse events for specific drugs, the models’ responses reflect the statistical weight of their training data, including patient narratives and published case reports.
Several academic groups have published studies using LLMs as pharmacovigilance tools — essentially asking large models to identify drug-adverse event pairs from their training data and comparing those pairs against known FAERS signals. A 2024 study in Nature Digital Medicine found that GPT-4 correctly identified 73 percent of established FAERS signals from prompting alone, without access to the underlying FAERS database. It also generated candidate signals for several drug-event pairs that had not yet reached statistical significance in FAERS but did so subsequently.
The implications are double-edged. On the benefit side, LLMs may function as early warning tools that distill signal from the entirety of published medical literature faster than any human literature review process. On the risk side, LLMs can also generate plausible-sounding but unvalidated drug-adverse event associations that circulate in patient communities and create nocebo effects — real symptoms driven by expectation rather than pharmacology. Distinguishing true pharmacovigilance signals from LLM-generated artifacts requires exactly the kind of human-AI hybrid review that platforms like DrugChatter are built to support.
Brand Share of Voice in the Age of AI Health Information
Pharmacovigilance and brand monitoring are not typically discussed in the same sentence. They should be. A drug’s reputation in AI-generated health information — the summary that ChatGPT returns when a patient asks about it, the recommendation that a symptom-checking app generates, the response that a hospital’s patient portal chatbot gives when asked about side effects — is shaping prescribing behavior in ways that traditional market research does not capture.
This is the brand share of voice problem for the AI age. For the previous two decades, share of voice was measured in digital advertising impressions, journal publication counts, speaker bureau reach, and HCP contact frequency from sales representatives. All of those measures are still relevant. None of them captures what AI systems say about a drug when no brand team is in the room.
What AI Systems Say About Your Drug
The variation in what different AI systems say about the same drug is substantial and consequential. An analysis conducted by DrugChatter in Q1 2026 compared the responses of five major AI health information systems to standardized queries about five drugs in the GLP-1 receptor agonist class. The variation in adverse event characterization, dosing information accuracy, and comparative positioning was significant enough that a prescriber using different AI tools to inform a treatment decision would receive materially different information about the same products.
Semaglutide (Ozempic/Wegovy, Novo Nordisk) received consistently more favorable characterization in terms of cardiovascular benefit language than tirzepatide (Mounjaro/Zepbound, Eli Lilly) across the AI systems tested, despite the fact that tirzepatide’s cardiovascular outcome data in the SURMOUNT-MMO trial, published in late 2024, showed comparable or superior outcomes on several endpoints. That discrepancy reflects training data lag — AI models trained on data through mid-2024 capture Novo Nordisk’s cardiovascular positioning more thoroughly than they capture Lilly’s more recent data.
For a pharmaceutical brand team, the actionable question is: what does the AI information environment say about our drug right now, and where does it diverge from our current label claims and approved promotional messaging? Answering that question requires systematic monitoring of AI outputs, not just traditional digital media.
Regulatory Risk from AI Misinformation
The regulatory risk from AI health misinformation flows in both directions. If an AI system overstates a drug’s benefits relative to its label — claiming, for example, that a drug is indicated for a condition it is not approved to treat — and a physician or patient acts on that claim, the pharmaceutical company that makes the drug faces potential liability even if it had nothing to do with training the AI system.
The FDA’s Division of Drug Information has received inquiries from multiple companies asking precisely this question: what is a company’s regulatory exposure when an AI system generates off-label promotional content about its product without the company’s involvement? The agency has not answered that question with formal guidance. It has signaled, through informal communications and presentations at public meetings, that companies should monitor AI-generated content about their products and report to the FDA any instances where that content contains significant safety misinformation.
That informal expectation — monitor AI content about your drug and report significant misinformation — is a new operational requirement for pharmaceutical brand and medical affairs teams. It did not exist in any meaningful form before 2023. It is now a real operational task that requires real infrastructure.
Building an AI Pharmacovigilance Operation That Holds Up in Court
The standards for a defensible pharmacovigilance operation, AI-assisted or otherwise, are well-established in regulatory guidance and case law. What changes with AI is the evidentiary bar: AI systems generate logs, timestamps, confidence scores, and audit trails that become discoverable in litigation. Building that infrastructure correctly from the start is less expensive than defending it after the fact.
Validation Requirements
Any AI tool used in the regulatory pharmacovigilance pipeline — case intake, MedDRA coding, signal detection — requires validation under the same Computer Software Assurance (CSA) framework the FDA articulated in its 2022 guidance replacing the older CSV approach. The practical difference between CSA and CSV for AI systems is significant: CSA is risk-based, meaning the validation burden scales to the criticality of the system’s function in the regulatory workflow.
For a system that performs automated case triage but has a human reviewer confirm each output before regulatory submission, the validation burden is lower than for a system that directly generates expedited reports. Companies deploying AI pharmacovigilance tools need to map each AI function against its position in the submission workflow and apply validation effort proportionately. The worst outcome — and a common one in FDA inspections — is deploying an AI tool that automates a regulatory-critical function without the validation documentation to prove it works reliably.
Human Oversight Requirements
Neither the FDA nor the EMA has accepted fully automated pharmacovigilance without human oversight for regulatory submissions. Every AI-assisted case processing workflow that produces a regulated output — an expedited report, a PADER, a signal assessment for a PSUR — requires documented human review at the output stage. AI systems function as force multipliers and triage tools; they do not yet function as autonomous regulatory agents.
The practical implication for staffing is not that AI pharmacovigilance reduces headcount to zero. It reduces the headcount required for data entry, MedDRA coding, and narrative drafting — the high-volume, lower-judgment tasks — while maintaining or increasing the demand for medical reviewers who can assess signal strength, contextualize benefit-risk, and make the judgment calls that determine what gets reported, when, and with what level of urgency.
Documentation and Audit Trail Design
The audit trail requirements for AI-assisted pharmacovigilance are more demanding than for manual workflows, not less. When a human safety associate makes a coding decision, the audit trail shows what they did. When an AI system makes a coding decision and a human confirms it, the audit trail must show what the AI recommended, what confidence level it assigned, what the human reviewer saw, and what decision the reviewer made. That fuller audit trail is discoverable in litigation and subject to inspection.
Companies that treat AI audit trails as a compliance checkbox rather than a strategic document are missing the legal value. A well-designed audit trail showing that an AI system flagged a signal, a human reviewer assessed it, and the company acted within a defined decision window is the strongest possible defense against a ‘knew or should have known’ claim. It demonstrates that the company operated the best available surveillance technology at the relevant time and responded to what that technology found.
DrugChatter: Tracking AI Mentions of Your Drug Across the Information Ecosystem
The pharmacovigilance infrastructure most large pharmaceutical companies have built is adequate for processing cases that arrive through defined channels: FAERS direct reporting, spontaneous reports from HCPs, call center intake, and clinical trial adverse event collection. It is less adequate — and in many cases entirely absent — for monitoring what AI systems say about drugs in real time.
DrugChatter fills that gap. The platform monitors drug mentions across AI-generated content, patient communities, social media, professional forums, and digital health applications. It uses natural language processing to identify content that meets the pharmacovigilance case threshold and separates pharmacovigilance-relevant content from general brand sentiment, competitive intelligence, and scientific discussion.
The competitive intelligence function is distinct from the pharmacovigilance function but operationally related. When AI systems consistently characterize a competitor’s drug more favorably than yours in patient-facing health information, that represents a share of voice problem that affects prescribing behavior. When those same AI systems mischaracterize your drug’s safety profile, that represents both a brand problem and a potential regulatory obligation. Both require monitoring. Both require a documented response workflow.
The Real-Time Signal Dashboard
DrugChatter’s core deliverable for pharmacovigilance teams is a real-time signal dashboard that aggregates AI-generated and user-generated mentions of a drug, scores them for pharmacovigilance relevance, and routes caseable content directly to the company’s case management system. The routing uses structured APIs that connect to Veeva Vault Safety, Oracle Argus, and IQVIA ARISg, the three dominant case management platforms, allowing the monitoring data to flow into the existing regulatory submission workflow without requiring a separate data entry step.
For brand and medical affairs teams, the dashboard provides share of voice tracking across AI health platforms — showing how ChatGPT, Perplexity, Google’s AI Overviews, and major health information AI tools characterize a drug relative to competitors in the same therapeutic class. That data feeds brand strategy, medical education planning, and the decision about when to approach an AI platform provider about correcting a material factual error in how their system characterizes a drug.
The Competitive Implications: Why This Is Also a Business Intelligence Problem
Pharmaceutical executives who think about AI pharmacovigilance only as a compliance function are underestimating its competitive implications. Safety signal timing is commercially consequential. A company that detects a comparative safety signal in a competitor’s drug before that signal reaches the regulatory record — and before the competitor’s label is updated — has information that affects formulary positioning, payer negotiations, and prescriber communications.
This is not hypothetical. The competitive dynamics around GLP-1 receptor agonist cardiovascular outcomes data played out in real time in 2024 and 2025, with competing companies watching each other’s trial readouts and real-world pharmacovigilance data for signals that could shift market share. The company that had better surveillance of what patient communities were saying about gastrointestinal tolerability — a key differentiator in that class — had an advantage in positioning its product before formal label updates reflected the emerging data.
Early Signal Detection as Market Intelligence
Real-world safety data has a competitive dimension that goes beyond the obvious. When post-marketing surveillance detects a safety signal in a competitor’s drug before that drug’s label is updated, a company’s medical affairs team can begin preparing comparative safety communications to HCPs. Those communications must stay within the bounds of approved promotional messaging and cannot rely on unapproved comparisons, but they can accurately characterize the known safety profile of both products as reflected in current labeling.
The company with better surveillance sees the signal first. It has more time to prepare its response. That time advantage translates into prescriber conversations that happen before the competitor’s label change, not after.
Formulary and Payer Strategy
Pharmacy benefit managers and health plan formulary committees increasingly incorporate real-world safety data into drug coverage decisions. A drug’s real-world adverse event profile, as reflected in post-marketing surveillance data, affects prior authorization criteria, step therapy protocols, and formulary tier placement. Companies that can present clean, well-documented real-world safety data to payers — data showing their product’s post-marketing profile is better than or consistent with clinical trial results — have a formulary negotiation advantage.
AI pharmacovigilance generates that data more rapidly and comprehensively than traditional surveillance methods. A company that monitors patient communities, social media, and AI-generated health content for adverse event signals has a richer real-world safety dataset than one that relies exclusively on FAERS submissions and HCP-reported cases. That dataset is a commercial asset, not just a compliance deliverable.
Implementation: What a Modern AI Pharmacovigilance Stack Looks Like
Building or procuring an AI-assisted pharmacovigilance operation requires decisions across four layers: data sources, AI capabilities, regulatory workflow integration, and governance. Most companies are not building from scratch — they are layering AI capabilities onto existing case management infrastructure. The layering decisions determine the system’s regulatory defensibility and operational effectiveness.
Data Sources
A complete data source strategy for AI pharmacovigilance covers four categories:
- Structured regulatory data: FAERS, EudraVigilance, MHLW’s JADER, and equivalent national databases, accessed via APIs or direct download and processed against the company’s own product portfolio
- Unstructured patient-generated content: social media, patient forums, health community platforms, and AI-generated health information outputs
- Electronic health records and claims data: through partnerships with real-world data aggregators such as IQVIA, Symphony Health, Optum, or through hospital system data sharing agreements
- Published literature: continuously monitored through NLP-assisted literature surveillance that flags new case reports and observational studies for medical review
AI Capabilities Required
The AI stack for pharmacovigilance monitoring requires capabilities across two domains:
- Case identification: NLP for adverse event extraction, named entity recognition for drug and patient identification, and classification models for case validity triage
- Signal detection: statistical disproportionality methods applied to structured case data, topic modeling for emerging themes in unstructured text, and time-series anomaly detection for changes in reporting frequency that may indicate a new signal
Regulatory Workflow Integration
AI outputs must connect to regulatory submission workflows without creating parallel data management processes that diverge from the system of record. The integration architecture should place AI-identified cases directly into the company’s validated safety database — not into a separate tracking system — where they undergo the same medical review and reporting assessment as traditionally-sourced cases. Parallel tracking creates exactly the kind of inconsistency that FDA inspectors target.
Governance: Who Owns the Signal
The organizational question that AI pharmacovigilance raises most sharply is who owns the signal. In traditional pharmacovigilance operations, the safety physician or medical officer makes the signal assessment and owns the decision to escalate or close. When AI systems are surfacing signals from social media, patient forums, and AI health content platforms, those signals arrive in a different operational context than a reported adverse event from a clinical trial or an HCP call.
Companies need clear governance protocols that define who receives AI-surfaced signals, what review process they undergo, what the decision criteria are for upgrading them to formal signal assessments, and how long the review window is before a signal must be either acted on or documented as closed. Absence of governance protocols means AI-surfaced signals accumulate without disposition, creating the exact ‘knew or should have known’ liability that the monitoring was supposed to prevent.
Where the Field Goes in the Next Three Years
The regulatory, technological, and competitive dynamics around AI pharmacovigilance are moving quickly enough that the standard of care in 2029 will look materially different from today’s. Three trends are worth tracking.
First, the FDA is likely to publish binding guidance on AI-assisted pharmacovigilance within the next 18 to 24 months. The agency’s 2024 AI action plan committed to specific pharmacovigilance guidance as a deliverable. When that guidance arrives, it will set minimum standards for AI tool validation, audit trail requirements, and human oversight levels that currently operating companies will need to meet. Companies that have already built AI-assisted operations will be better positioned to comply than those that treat the guidance as a starting gun.
Second, multimodal AI — systems that process text, audio, and image data together — will expand the pharmacovigilance data surface. Patient-generated video content about drug experiences is already substantial on TikTok and YouTube. Processing audio transcripts of those videos at scale for pharmacovigilance signals is technically feasible with current AI capabilities and operationally unexploited by most companies. The next generation of social listening platforms, including DrugChatter’s roadmap, incorporates audio and video processing alongside text.
Third, federated learning approaches will allow companies to train signal detection models on pooled pharmacovigilance data without sharing proprietary case data. Several academic consortia and industry groups, including TransCelerate BioPharma and the Reagan-Udall Foundation, are exploring federated pharmacovigilance models that would allow industry-wide signal detection on a scale no single company’s database can support. Those models, when operational, will detect cross-industry signals — events that appear distributed across multiple companies’ surveillance systems but that aggregate into a clear signal when pooled — faster than any individual company’s system can.
Key Takeaways
- Traditional pharmacovigilance captures roughly 60 to 70 percent of a drug’s adverse event profile from clinical trials. AI-assisted post-marketing surveillance closes part of the remaining gap — and the companies that close it fastest face lower litigation exposure.
- The FDA and EMA have not finalized AI-specific pharmacovigilance guidance, but both agencies are signaling that formal standards are coming. The validation and audit trail requirements they will impose are predictable enough to build toward now.
- Social media monitoring for pharmacovigilance purposes creates regulatory obligations. A company that monitors social media for marketing and simultaneously encounters caseable adverse event content cannot claim ignorance. The audit trail is the protection.
- AI-generated health content is now a distinct pharmacovigilance data stream. What chatbots and AI health platforms say about drugs reaches patients at scale and requires systematic monitoring for both adverse event case generation and safety misinformation.
- The ‘knew or should have known’ legal standard shifts as AI detection capabilities improve. A company operating below the current standard of care in pharmacovigilance faces higher litigation exposure than it did a decade ago, because the state-of-the-art capability it failed to deploy is no longer experimental.
- Brand share of voice in AI health information is measurable and commercially consequential. Companies that monitor what AI systems say about their drugs — and about competitors’ drugs — have an intelligence advantage in formulary negotiation, prescriber communications, and competitive positioning.
FAQ
Does monitoring social media for adverse events create a legal obligation to report everything that appears there?
Not everything, but more than most companies report today. The four-element test — a patient identifier, a reporter, a suspect drug, and an adverse event — determines whether content constitutes a case that must be processed and, if serious and unexpected, reported within 15 days. Social media posts frequently contain all four elements, even when the poster does not intend to file a formal adverse event report. The practical standard is: if a company monitors a channel and an employee or automated system reviews content from that channel, the company has ‘received’ what is in that content for regulatory purposes. Selective processing — reviewing for brand mentions but ignoring adverse events on the same platform — is not a defensible compliance position if the adverse event was visible in the same data stream the company was already monitoring.
What is the regulatory status of using large language models for MedDRA coding in regulatory submissions?
There is no specific FDA or EMA guidance that either approves or prohibits LLMs for MedDRA coding in regulatory submissions, as of Q2 2026. Companies are operating under general Computer Software Assurance principles, which require risk-based validation of any software used in regulated workflows. LLM-based coding tools are being used in production environments by several large pharmaceutical companies, with human review as the final step before submission. The practical regulatory risk is not in using the technology but in inadequate validation documentation — specifically, the absence of performance metrics showing the model’s accuracy on the specific case types and terminology the company encounters in its portfolio.
How does AI pharmacovigilance affect a company’s exposure in product liability litigation?
It cuts both ways. Earlier signal detection, followed by timely label updates and safety communications, reduces exposure by shortening the window in which patients are harmed without adequate warning. But deploying AI pharmacovigilance also raises the standard of care: a company that has the capability to detect signals earlier is expected to act on what its systems find. An AI system that flagged a signal that was then not escalated or acted upon creates a discoverable record of inaction that is more damaging in litigation than no record at all. The liability management value of AI pharmacovigilance depends entirely on the governance processes that act on what the AI finds.
What makes AI-generated drug misinformation a pharmacovigilance issue rather than just a brand communications problem?
The distinction matters when AI-generated content causes patients to behave differently with respect to a drug in ways that produce adverse outcomes. If an AI health platform tells patients that a specific drug has no serious drug interactions when the label documents several, and a patient experiences a serious adverse event from a drug interaction that the AI’s inaccurate information allowed to happen, the adverse event report that follows is a pharmacovigilance case — not just a brand problem. The pharmaceutical company is not liable for what the AI system said, but it does have a regulatory obligation to process the resulting adverse event case and to report the AI misinformation to the FDA if the misinformation is causing a pattern of safety-relevant patient behaviors. That line between brand problem and pharmacovigilance obligation crosses when AI content generates cases.
What is the practical ROI calculation for investing in AI-assisted pharmacovigilance versus expanding traditional surveillance headcount?
The cost comparison is straightforward for case processing volume: AI-assisted intake reduces the cost per case by 40 to 60 percent for high-volume case types, based on benchmarking data from ZS Associates and IQVIA. At 100,000 cases per year — a mid-size company’s volume for a major marketed product — that reduction represents $8 to $15 million annually in operational savings at typical fully-loaded case processing costs. The litigation avoidance value is harder to calculate precisely but is substantially larger for any company with a marketed product in a class with active plaintiff’s bar attention. The more compelling ROI argument is the asymmetric one: the cost of deploying AI pharmacovigilance is fixed and predictable; the cost of missing a signal that AI would have caught is not.





